THE FEMALE HEALTH COMPANY
FORM 10-K
September 30, 2016
|
PART I |
Page |
|||
|
|
||||
|
|
5 |
|
||
|
|
21 |
|
||
|
|
36 |
|
||
|
|
36 |
|
||
|
|
36 |
|
||
|
|
36 |
|
||
|
|
|
|
|
|
|
PART II |
|
|
|
|
|
|
|
|
|
|
|
|
37 |
|
||
|
|
39 |
|
||
|
Management's Discussion and Analysis of Financial Condition and Results of Operations |
|
40 |
|
|
|
|
47 |
|
||
|
|
47 |
|
||
|
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
|
47 |
|
|
|
|
47 |
|
||
|
|
47 |
|
||
|
|
|
|
|
|
|
PART III |
|
|
|
|
|
|
|
|
|
|
|
|
48 |
|
||
|
|
48 |
|
||
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
48 |
|
|
|
Certain Relationships and Related Transactions, and Director Independence |
|
49 |
|
|
|
|
49 |
|
||
|
|
|
|
|
|
|
PART IV |
|
|
|
|
|
|
|
|
|
|
|
|
50 |
|
||
|
|
|
54 |
|
|
3
FORWARD-LOOKING STATEMENTS
Certain statements included in this Annual Report on Form 10-K which are not statements of historical fact are intended to be, and are hereby identified as, "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements can be identified by the use of forward-looking words or phrases such as "anticipate," "believe," "could," "expect," "intend," "may," "opportunity," "plan," "predict," "potential," "estimate," "will," "would" or the negative of these terms or other words of similar meaning. These statements are based upon the Company's current plans and strategies, and reflect the Company's current assessment of the risks and uncertainties related to its business, and are made as of the date of this report. The Company cautions readers that forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause the actual results, performance or achievements of the Company to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Such factors include, but are not limited to, those described under the caption "Risk Factors" in Item 1A. of this report. The Company undertakes no obligation to make any revisions to the forward-looking statements contained in this report or to update them to reflect events or circumstances occurring after the date of this report.
4
PART I
General
The Female Health Company is a medical therapeutics company, with an initial focus on the development and commercialization of pharmaceuticals for men’s and women’s health and oncology that qualify for the U.S. Food and Drug Administration's (FDA) 505(b)(2) accelerated regulatory approval pathway. The Company also has a Consumer Health and Medical Devices Division and Global Public Health Sector Division. The Company does business as both "Veru Healthcare" and "The Female Health Company." The Company is organized as follows:
|
· |
Veru Healthcare manages: |
|
o |
The Pharmaceuticals Division, which develops and commercializes pharmaceutical products for men's and women's health and oncology. |
|
o |
The Consumer Health and Medical Devices Division, which is focused on commercializing sexual healthcare products and devices for the consumer market, including the Company’s Female Condom (FC2) for over-the-counter (OTC), and as the Female Disposable Contraceptive Device (FC2) in the U.S. prescription market, as well as PREBOOST® (benzocaine 4%) medicated individual wipes which is a male genital desensitizing drug product that helps in the prevention of premature ejaculation. |
|
· |
The Female Health Company manages the Global Public Health Sector Division, which is focused on the FC2 Female Condom® in the global public health sector business. This division markets FC2 to public health entities, including ministries of health, government health agencies, U.N. agencies, nonprofit organizations and commercial partners, that work to support and improve the lives, health and well-being of women around the world. |
On October 31, 2016, as part of the Company's strategy to diversify its product line to mitigate the risks of being a single product company, the Company completed a merger transaction (the APP Merger) with Aspen Park Pharmaceuticals, Inc. (APP). APP is a medical therapeutics company focused on the development and commercialization of pharmaceutical and consumer health products for men's and women's health and oncology. For men, product and product candidates are in the areas of benign prostatic hyperplasia, male infertility, amelioration of side effects of hormonal prostate cancer therapies, gout, sexual dysfunction, and prostate cancer. For women, product candidates are for advanced breast and ovarian cancers and for female sexual health.
On August 12, 2016, the FDA agreed that the Company's Tamsulosin DRS product, a proprietary medication for the treatment of benign prostatic hyperplasia (BPH), a $3.5 billion market, qualifies for the accelerated 505(b)(2) regulatory approval pathway and with APP's plans to conduct a single bioequivalence study to support the filing of a new drug application (NDA). The Company plans to initiate a bioequivalence clinical study by the first quarter of 2017, submit an NDA for Tamsulosin DRS in 2017 and, if approved, launch the product in early 2018.
On October 31, 2016, the Company completed an interim analysis of the double-blind, randomized placebo controlled clinical trial of its novel PREBOOST® product. The Company plans to launch PREBOOST® in the United States before the end of 2016.
The Company accepted an invitation from the FDA to present at the meeting of the Bone, Reproductive and Urologic Drugs (BRUD) Advisory Committee on December 6, 2016. The FDA uses advisory committees to obtain independent expert advice on scientific, technical and policy matters. At the meeting, the committee discussed appropriate clinical trial design features, including acceptable endpoints for demonstrating clinical benefit, for drugs intended to treat secondary hypogonadism (low testosterone levels) while preserving or improving testicular function, including spermatogenesis. At the meeting, the FDA Advisory Committee provided guidance for clinical trial design and endpoints. The committee agreed with the intended patient population to treat, recommended a short-term study, and supported the use of improvement of semen quality for such clinical endpoints as avoidance of aggressive assisted reproductive procedures such as in vitro fertilization or pregnancy. Based on this advice, the Company plans to file an investigational new drug application (IND) in 2017 and advance MSS-722 into Phase 2 clinical trial in men with testicular dysfunction [severe oligospermia (low sperm count) and secondary hypogonadism] as a cause of male factor infertility.
Prior to the completion of the APP Merger, the Company had been a single product company, focused on manufacturing, marketing and selling FC2. FC2 is the only currently available female-controlled product approved for market by the FDA and cleared by the World Health Organization (WHO) for purchase by U.N. agencies that provides dual protection against unintended pregnancy and sexually transmitted infections (STIs), including HIV/AIDS and the Zika virus.
5
The Company currently operates in one industry segment which includes the development, manufacture, and marketing of consumer health care products. Therefore, no segment data is disclosed in the Notes to the Consolidated Financial Statements contained in this report. Information regarding the Company's operations by geographic area is included in Note 10 in the Notes to the Consolidated Financial Statements contained in this report.
Company History
The Female Health Company is the successor to The Wisconsin Pharmacal Company, Inc. (Wisconsin Pharmacal), a company which manufactured and marketed disparate specialty chemical and branded consumer products. Wisconsin Pharmacal was originally incorporated in 1971.
The FDA approved the Company's first generation Female Condom, FC1, for distribution in the U.S. in 1993 and approved the Company's U.K. FC1 manufacturing facility in 1994. Prior to 1996, Wisconsin Pharmacal owned certain rights to the Female Condom in the U.S., Canada, and Mexico. In 1996, the Company completed a series of actions which resulted in the Company's acquisition of worldwide rights to FC1, the divestiture of Wisconsin Pharmacal's other businesses and the change of the Company's name to "The Female Health Company." As a result of these actions, the Company's sole business consisted of the manufacture, marketing, and sale of the FC1 Female Condom.
In 2005, the Company completed the development of its second generation Female Condom (FC2). FC2 was first marketed internationally in March 2007 and has been marketed in the U.S. since August 2009. FC2 was approved by the FDA as a Class III medical device on March 10, 2009. In addition to FDA approval, FC2 has been approved by other regulatory agencies, including the European Union, India, and Brazil. Based on a rigorous scientific review, WHO cleared FC2 for purchase by U.N. agencies in 2006.
On October 31, 2016, the Company completed the APP Merger. Pursuant to the APP Merger, the outstanding shares of APP common stock and preferred stock were converted into the right to receive in the aggregate 2,000,000 shares of FHC's common stock (the FHC Common Stock) and 546,756 shares of FHC Class A Convertible Preferred Stock - Series 4 (the Series 4 Preferred Stock). After giving effect to the conversion of the Series 4 Preferred Stock to FHC Common Stock, which is wholly dependent upon future shareholder approval, the former APP stockholders will own 23,870,249 shares of FHC Common Stock in total, constituting approximately 45% of the outstanding shares of FHC Common Stock as of October 31, 2016. The total estimated purchase price of approximately $22,676,737 is based on the closing price of FHC Common Stock of $0.95 per share on October 31, 2016 and the issuance to the APP stockholders of a total of 23,870,249 shares of FHC Common Stock. The Company is currently in the process of determining the fair value of the assets acquired and liabilities assumed in the business combination.
Strategy
Our goal is to be a leader in men's and women's health and oncology by developing a portfolio of pharmaceutical products that address significant health needs in large potential markets. We have combined the revenue and cash flows from the market leading FC2 female condom with APP's deep pipeline of pharmaceutical and consumer health product candidates. Initially, we intend to focus on the three low-cost, near-term and potentially high-reward programs that are expected to qualify for the abbreviated 505(b)(2) FDA regulatory pathway: Tamsulosin DRS for BPH, MSS-722 for male infertility, and APP-944 for hot flashes in men taking hormonal therapies for advanced prostate cancer. The 505(b)(2) regulatory pathway can result in a much less expensive and faster route to approval, compared with the traditional 505(b)(1) regulatory development path, while creating new, differentiated products with potentially high commercial value.
The key elements of our strategy are as follows:
|
· |
Obtain regulatory approvals of products in North America, Europe and Asia. Assuming the successful completion of clinical trials, we expect to file, or a partner will file on our behalf, for regulatory approval of our pharmaceutical products in North America, Europe and Asia, including Tamsulosin DRS for the treatment of BPH, MSS-722 for the treatment of male infertility, APP-944 for the treatment of hot flashes in men on prostate cancer hormonal therapies, APP-111 for the treatment of metastatic prostate, breast and ovarian cancers and APP-111/112 for the prevention and treatment of gout and Familial Mediterranean Fever (FMF). |
6
|
· |
Develop a portfolio of men's and women's health and oncology products. We have developed or acquired development and marketing rights to a portfolio of men's and women's health and oncology products and intend to continue to acquire, in-license and develop new products that we believe offer unique market opportunities and/or complement existing product lines. We have adopted a three-tier strategy with respect to licensing or acquiring new products and technologies designed to diversify the risks inherent in traditional pharmaceutical development: (i) license or acquire fully-developed, FDA-approved products that have development potential and offer certain market protection against competitors, such as patent rights, marketing exclusivity or orphan drug designation; (ii) create differentiated products with potentially high commercial value by selecting a new indication for or modifying existing FDA-approved products utilizing the 505(b)(2) FDA approval pathway; and (iii) identify and acquire products and technologies in late preclinical or early clinical stages of development to minimize the time and expense of development. |
|
· |
Focus on products with significant potential commercial opportunities in men's and women's health and oncology markets. We intend to focus on developing drugs that we believe have potential significant commercial opportunities in markets. The core areas of interest include BPH, sexual dysfunction, prostate, breast and ovarian cancer therapies, amelioration of prostate cancer hormonal therapy side effects, male infertility, gout and FMF. We believe that these areas of the pharmaceutical market are large, growth markets. Through continued specialization as a men's and women's health and oncology company and by continuing to refine its capabilities in clinical research and development and marketing, we believe we can develop a strong position to be a leader in these markets. |
|
· |
Develop business and enhance research through strategic alliances. A key component of our business strategy is to leverage the resources gained from each collaboration to expand our technology and operations base. In addition, we believe collaborations with academic centers and small discovery innovative companies will supplement the scientific resources available to us and broaden access to rapidly emerging drug discovery candidates. |
|
· |
Develop opportunities in the consumer and prescription markets. The Company believes that there are opportunities to develop the prescription market for FC2 as a female disposable contraceptive device, and that such marketing of FC2 will complement the consumer launch of PREBOOST®. The Company recently appointed a Vice President of Marketing for Veru Healthcare to oversee the implementation of its marketing plan for FC2 by prescription and PREBOOST®, as well as the future pre-launch and launch activities for Tamsulosin DRS. |
|
· |
Continue efforts in the global public sector. The Company intends to continue to develop global markets for FC2 for both contraception and STI prevention, including HIV/AIDS and the Zika virus. The Company has developed contacts and relationships with global public health sector organizations such as WHO, UNFPA, USAID, and the United Nations Joint Programme on HIV/AIDS (UNAIDS), country-specific health ministries, NGOs and commercial partners in various countries. The Company has recently appointed a President for the Global Public Health Sector Division of The Female Health Company and also has representatives in various locations around the world to provide technical and marketing support as well as assist with its customers’ prevention and family planning education programs. |
|
· |
Capitalize on expertise and reputation of our management team and scientific advisors. Our management has significant expertise and experience in men's and women's health, urology and oncology as well as drug development, marketing and sales which will enable us to manage effectively the preclinical studies and clinical trials of drug candidates and product commercialization. In addition, we intend to capitalize on the strong reputations of the members of our management and board of directors with academic institutions, hospitals, physicians, pharmacists and distributors to expand its customer base and to introduce new products. |
Products
The following table summarizes the current status of the Company’s product portfolio:
|
|
|
U.S. |
|
|
|
|
|
REGULATORY |
DEVELOPMENT |
|
|
PRODUCT |
INDICATION |
PATHWAY |
PHASE |
|
|
|
|
|
|
|
|
Pharmaceuticals Division |
|
|
|
|
|
|
|
|
|
|
|
Tamsulosin Delayed Release Sachet (DRS) (tamsulosin HCl for extended-release oral suspension) |
Benign prostatic hyperplasia |
505(b)(2) |
Bioequivalence study |
|
|
|
|
|
|
|
|
MSS-722 (Fixed ratio of trans- and cis-clomiphene citrate isomers) |
Male infertility caused by testicular dysfunction |
505(b)(2) |
Phase 2 |
|
7
|
|
|
|
|
|
|
APP-944 (Zuclomiphene citrate) |
Hot flashes in men on prostate cancer hormonal therapies |
505(b)(2) |
Phase 2 |
|
|
APP-111- Oral tubulin targeting chemotherapy |
Metastatic prostate, breast, and ovarian cancers |
505(b)(1) |
Preclinical |
|
|
APP-111/112- Oral agent that targets colchicine binding site of tubulin |
Gout and Familial Mediterranean Fever |
505(b)(1) |
Preclinical |
|
|
Consumer Health and Medical Devices Division |
|
|
|
|
|
|
|
|
|
|
|
PREBOOST® (4% benzocaine wipes) |
Premature ejaculation |
FDA monograph compliant |
Marketed |
|
|
Female disposable contraceptive device (FC2) for prescription |
Unintended pregnancy and STIs |
FDA approved |
Marketed |
|
|
FC2 Female Condom® |
Unintended pregnancy and STIs |
FDA approved |
Marketed |
|
|
|
|
|
|
|
|
Global Public Health Sector Division |
|
|
|
|
|
FC2 Female Condom® |
Unintended pregnancy and STIs |
FDA approved |
Marketed |
|
|
|
|
|
|
|
Pharmaceutical Product Candidates
Tamsulosin DRS (tamsulosin HCl for extended-release oral suspension) for the treatment of BPH
Scientific Overview. Tamsulosin DRS is a new slow release granules formulation containing the active pharmaceutical ingredient in FLOMAX® (tamsulosin HCl) capsules which is a commonly used medicine for the treatment of BPH, also known as enlargement of the prostate. FLOMAX® is indicated for the treatment of the signs and symptoms of BPH. Tamsulosin is a selective alpha1 adrenergic receptor blocking drug that is specific for the alpha1 adrenergic receptors located in the smooth muscle of the prostate and bladder neck. Symptoms associated with BPH occur because of increased smooth muscle tone of the prostate and bladder which leads to constriction of urinary flow, urinary retention, urinary infection, kidney damage and life threatening blood infection called urosepsis. Blocking these alpha1 adrenergic receptors relaxes the smooth muscles of the prostate and bladder neck resulting in the reduction in the symptoms of BPH and improvement of urinary flow rate. FLOMAX® capsules should not be crushed, chewed or opened as is stated in the FDA package insert, because it cannot be reliably absorbed into the bloodstream. As a consequence, men will have drug levels that will not treat their BPH and are placed at higher risk for postural hypotension (sudden drop in blood pressure upon standing that can lead to fainting). Tablets and capsules are problematic for 15% of men over the age of 60 in the general community and the up to 60% of men in long term facilities who have difficulty swallowing tablets and capsules because of certain medical conditions, including degenerative neurological diseases like Parkinson's or having suffered a stroke. Not being able to take alpha blocker drugs for BPH, like FLOMAX®, because of difficulty swallowing tablets and capsules may lead to the increased risk of acute urinary retention, urinary catheterization, urosepsis and death. Because Tamsulosin DRS is a new proprietary slow release granules formulation containing the active pharmaceutical ingredient in FLOMAX®, it would provide a more convenient and reliable way to deliver therapeutic levels of tamsulosin to men who have difficulty swallowing tablets and capsules.
Development Plan. This new formulation called Tamsulosin DRS contains the same tamsulosin active pharmaceutical ingredient that is found in FLOMAX® (tamsulosin HCI) capsules and, as such, would be expected to have the same efficacy and safety as FLOMAX®. This information can be referenced under a 505(b)(2) NDA submission for Tamsulosin DRS. On August 12, 2016, the FDA cleared Tamsulosin DRS for the accelerated 505(b)(2) regulatory approval pathway and agreed with our plans to conduct a single bioequivalence study to support the filing of an NDA. The Company plans to initiate a three-week bioequivalence study by the first quarter of 2017, submit an NDA for Tamsulosin DRS in 2017 and, if approved, launch the product in early 2018.
Market. The initial marketing plan will target men in long term care facilities and men in the community that have difficulty swallowing tablets and capsules. Initially, a sales force is not required for this product as pharmacists and physicians have the ability to identify and to provide the appropriate formulation of tamsulosin for a patient who has BPH and difficulty swallowing tablets and capsules. Based on IMS data, FLOMAX® and generic tamsulosin sales from March 2014 to March 2015 were $3.5 billion in the U.S. The U.S. market for all alpha blockers for BPH is estimated to be $4.5 billion annually per IMS. Men in long term care or nursing homes have up to a 60% prevalence of swallowing difficulties and account for about 13% of total tamsulosin sales, whereas over 15% of men over 60 years of age in the general population have difficulty swallowing tablets and capsules.
8
MSS-722 (Fixed ratio of trans- and cis- clomiphene citrate isomers) for the treatment of male infertility
Scientific Overview. Up to 10% of infertile men have an endocrine cause and 2% of infertile men have an adult onset form of idiopathic hypogonadotropic hypogonadism. Current FDA-approved treatments for this indication include Human Chorionic Gonadotropin (HCG) and Follicle Stimulating Hormone (FSH) injections. There are no FDA-approved oral therapies for male infertility. CLOMID (clomiphene citrate) 50mg tablets are being used off-label as first line empiric therapy in 90% of idiopathic infertile men. CLOMID is FDA-approved for the treatment of ovulatory dysfunction in women desiring pregnancy. CLOMID is a mixture of two geometric isomers cis-clomiphene (zuclomiphene) and trans-clomiphene (enclomiphene) containing between 30-50% of the cis-clomiphene isomer. Trans-clomiphene has antiestrogenic activity, while the cis-clomiphene has estrogenic activity. In men, clomiphene has the ability interact with the hypothalamus and pituitary gland to cause the secretion of Luteinizing Hormone (LH), and the higher levels of LH will stimulate Leydig cells in the testes to produce testosterone, to promote spermatogenesis, and to improve sperm count and quality.
Based on the scientific literature, clomiphene has demonstrated the ability to improve sperm quality and sperm counts in infertile men and result in higher pregnancy rates. Based on 39 published studies, clomiphene appears to be well tolerated in men with doses as high as 400 mg/day and up to three years of use. However, the efficacy results for an individual patient have been inconsistent from study to study for several reasons: the form of clomiphene used contains varying ratios of the trans- and cis-clomiphene isomers, different doses were given, various dosing schedules were followed and different patient populations were studied. Clomiphene has not been formally studied for regulatory approval for the indication of male infertility; therefore, there is no established dose or schedule for efficacy or safety in men. MSS-722 is a patented, proprietary daily oral tablet that has a specific fixed ratio of the combination of trans- and cis-clomiphene isomers.
Development Plan. MSS-722 is being developed as the first FDA-approved oral agent for the treatment of male infertility. MSS-722 has fixed ratio of the combination of trans- and cis- clomiphene isomers. We believe that using a fixed ratio approach will allow the determination of the correct dose and schedule for efficacy and safety for the treatment of male infertility. The patient population will be men who have hypogonadotropic hypogonadism and oligozoospermia (low sperm count). We met with the FDA for a pre-IND meeting on May 28, 2015 where the FDA confirmed that MSS-722 qualifies for the 505(b)(2) regulatory pathway. The formulation, doses and dosing regimen for MSS-722 will differ from those of CLOMID. Despite the differences, the approval of MSS-722 will rely on nonclinical and clinical efficacy and safety information from the listed drug labeling and in the published literature. On December 6, 2016, the Company was invited to discuss our clinical trial design and plans with the FDA as part of The Bone, Reproductive, and Urologic Drugs Advisory Committee Meeting. Based on positive regulatory recommendations by the BRUD Advisory Committee, we plan to file an IND and possibly initiate a Phase 2 clinical study in 2017. We have also filed with the FDA a request for orphan drug status on January 7, 2016. Orphan drug status, if granted by the FDA, will provide several regulatory benefits including seven years of market exclusivity.
Market. If approved, MSS-722 will be indicated as the first oral treatment for male infertility. Infertility affects 6.1 million couples in the United States representing 15% of all couples trying to conceive. Up to 50% of infertility is attributed to males who are subsequently found to have abnormal semen analysis, of which 50% of these men are diagnosed with idiopathic, or unexplained, infertility. Ninety percent of men with idiopathic male infertility are empirically treated with off-label use of CLOMID. MSS-722 may be effective in treating male factor infertility caused by testicular dysfunction [low sperm concentration (oligozoospermia) and low testosterone blood levels (hypogonadotropic hypogonadism)]. The current U.S. market size for male infertility is estimated to be $700 million annually based on current off-label use of CLOMID and clomiphene generics prescription data (IMS).
APP-944 (zuclomiphene citrate) for the treatment of hot flashes caused by prostate cancer hormonal therapies in men with advanced prostate cancer.
Scientific Overview. Prostate cancer is the most common noncutaneous cancer diagnosed in men, with over 200,000 new cases in the U.S. in 2016. The estimated prevalence of prostate cancer in the U.S. is 2.35 million cases for which over one-third will have received androgen deprivation therapy. Hot flashes, also known as vasomotor symptoms, are the most common and distressing side effect of prostate cancer hormonal therapies. Hormone therapies include androgen deprivation, like LUPRON (leuprolide) or ZOLADEX (goserelin), as well as the newer agents approved to treat advanced prostate cancer such as ZYTIGA (abiraterone) and XTANDI (enzalutamide). Up to 80% of men on androgen deprivation therapy complain of hot flashes. Hot flashes are defined as intense heat sensation, flushing and profuse sweating and chills as well as anxiety and palpitations. Although episodes of hot flashes occur repeatedly and last a few minutes, some may last up to 20 minutes. Hot flashes associated with prostate cancer hormonal therapies tend to persist over time with the same frequency and intensity throughout therapy. Up to 50% of men continue to report hot flashes after five years on prostate cancer hormonal therapy. Patients on prostate cancer hormonal therapy report significant effects on daily functioning and quality of life. Hot flashes are the main reason for patients to be noncompliant with their prostate cancer hormonal therapy. As prostate cancer patients with advanced and metastatic disease are living longer because of more effective hormonal therapies, hot flashes have become an even bigger concern and impact on quality of life.
9
Hormonal and nonhormonal therapies have been used off-label to treat hot flashes in men on prostate cancer hormonal therapies. In general, hormonal agents especially estrogens are effective at treating hot flashes. However, estrogen treatment is complicated by lack of consistent dosing, dose dependent gynecomastia (breast enlargement), gynecodynia (painful breasts) and increase in thromboembolic events. Nonhormonal agents that have been used off-label include anti-seizure agents and antidepressants that have bothersome side effects. Moreover, non-hormonal agents tend to be less efficacious than hormonal therapies for the treatment of hot flashes. There are no FDA-approved therapies for hot flashes caused by prostate cancer hormonal therapy in men with advanced prostate cancer. CLOMID (Clomiphene citrate), which contains 30-50% zuclomiphene, appears to be well-tolerated in 39 published studies in over 2,200 men with doses as high as 400 mg/day and up to three years of use. CLOMID (clomiphene citrate) contains the trans-isomer, enclomiphene which causes hot flashes. APP-944 is pure cis-clomiphene (zuclomiphete). Zuclomiphene is a potent nonsteroidal estrogen receptor agonist. We believe that a nonsteroidal hormone therapy like APP-944 has the potential to be an effective and well-tolerated treatment for hot flashes caused by prostate cancer hormonal therapies in men with advanced prostate cancer.
Development Plan. As APP-944, zuclomiphene, comprises 30-50% of CLOMID (clomiphene citrate) which is approved for the treatment of ovulatory dysfunction in women desiring pregnancy, the Company believes that it will be able to reference on nonclinical and clinical safety information from both the listed drug labeling and the published literature under the 505(b)(2) regulatory pathway. The Company plans to have a pre-IND meeting with the FDA in 2017, anticipates filing the IND in 2017 and plans to initiate a Phase 2 clinical study in early 2018.
Market. Hot flashes are the most common side effect of prostate cancer hormone therapy occurring in up to 80% of men, with about 30% having moderate to severe hot flashes. Approximately 700,000 men annually in the United States are on androgen deprivation therapy, abiraterone or enzalutamide for advanced prostate cancer. There are currently no FDA-approved therapies for hot flashes associated with prostate cancer hormonal therapies. The annual U.S. market for the treatment of hot flashes in men on prostate cancer hormonal therapies is estimated to be $600 million per IMS.
APP-111, a novel oral tubulin targeting chemotherapy, for the treatment of metastatic prostate cancer as well as metastatic breast and ovarian cancers.
Scientific Overview. In 2016, there were approximately 233,000 new cases of prostate cancer in the U.S. and about 25% will die from the disease. In the U.S., 5% of men with prostate cancer will have metastatic cancer and up to 30% of men with high-risk, localized prostate cancer will develop metastatic cancer following initial therapy. The median survival of patients with metastatic prostate cancer ranges from 3.2-4.5 years. For these men, the 1st line therapy is androgen deprivation therapy, or medical castration. Although most will initially respond, nearly all these patients will progress to metastatic castration resistant prostate and have a poor prognosis with an average survival of 1.5 years. New 2nd line hormonal agents, like XTANDI (enzalutamide) and ZYTIGA (abiraterone/prednisone) have resulted in an additional four to five months of average survival, but again, nearly all men on these agents will develop progressive metastatic prostate cancer.
Agents that target tubulin have been shown to be the most effective cytotoxic chemotherapy for the treatment of metastatic prostate cancer. Tubulin, a component of microtubules, is required for cancer cell replication and to shuttle the androgen receptor into the nucleus where the receptor stimulates genes for cancer cell proliferation. Docetaxel and cabazitaxel are examples of FDA-approved chemotherapy drugs that are given intravenously (IV) that target tubulin to treat metastatic prostate cancer. Although effective, the challenges for this class of chemotherapy agents, also known as taxanes, include that they must be given intravenously (IV) and that the cancer cells develop resistance to taxanes in a variety of ways: Cancer cells may (i) express multidrug resistance proteins which pump the taxane chemotherapy meant to kill the cancer cells, out of the cancer cells; (ii) develop tubulin mutations so taxanes are no longer able to bind to the mutated tubulin; and/or (iii) overexpress beta-tubulin so that there is plenty of tubulin present for cell replication even if some tubulin is bound by taxanes. There are also serious safety concerns with IV taxanes which include serious hypersensitivity reactions, myelosuppression and neurotoxicity such as peripheral neuropathy and muscle weakness.
Based on over 28 peer-reviewed scientific publications, APP-111 is a novel small molecule that is a new chemical entity (NCE) that has been optimized to be an orally dosed tubulin targeting chemotherapy agent. APP-111 binds to a different site from taxanes on tubulin called the "colchicine binding site." APP-111 has high oral bioavailability; does not interact with multiple drug resistance proteins so it cannot be pumped out of the cancer cell; minimal drug to drug interactions especially not metabolized by CYP3A4 and has high activity against many tumor types including prostate, breast and ovarian cancers. Furthermore, it has activity against cancers that have become resistant to taxanes, vinca alkaloids and doxorubicin. In preclinical studies, APP-111 has less neurotoxicity and leukopenia compared to other tubulin targeting agents.
10
Development Plan. The Company initially plans to develop APP-111 as a 3rd line hormonal therapy after androgen deprivation (1st line) and enzalutamide or abiraterone (2nd line) have failed. Production of the active pharmaceutical ingredient and preclinical safety toxicology studies required for an IND are expected to be completed in 2017, anticipate filing IND in late 2017 or early 2018, and Phase 1a and Phase 1b studies are planned for 2018. Phase 1 studies of APP-111 are planned in men who have metastatic prostate cancer that has progressed while taking androgen deprivation therapy and enzalutamide. After Phase 1s are completed, we plan to not only conduct a Phase 2 of APP-111 in men as 3rd line hormonal therapy in 2018, but also to start Phase 1s for metastatic breast and ovarian cancers in 2019.
Market. In the U.S., there is a $5 billion annual market for 2nd line hormone therapies for prostate cancer and a $4.8 billion annual market for IV-given taxanes and vinca alkaloids (docetaxel $1 billion and cabazitaxel $500 million in prostate cancer) per Decision Resources Group and Allied Market Research. Second line therapies like enzalutamide and abiraterone/prednisone have almost complete cross-resistance and should not be used in sequence for the treatment of metastatic prostate cancer. APP-111, as an oral tubulin targeting chemotherapy agent, could replace docetaxel and cabazitaxel. APP-111 could also be developed a 1st line therapy given with androgen deprivation in men who have hormone sensitive, high volume prostate cancer where androgen deprivation therapy and docetaxel have been shown in several studies to increase survival in these men by 17-21 months. Another 1st line indication could be developed in men who have metastatic prostate cancer and a mutation of the androgen receptor known as AR-V7. Prostate cancer hormone therapies are not effective in men who have AR-V7 mutations. However, this type of cancer appears to respond to docetaxel and may be potentially treated by an oral tubulin chemotherapy like APP-111. APP-111 could also be developed as an oral dosing alternative for the treatment of metastatic breast and ovarian cancers as these tumors also respond to IV taxanes.
APP-111 and APP-112 for the treatment of gout and Familial Mediterranean Fever.
Scientific Overview. Colchicine is FDA-approved for prophylaxis and treatment of gout flares in adults (0.6-1.2mg/day) and for FMF in adult and children four years and older (0.3-2.4mg/ day depending on age). Gout is a type of arthritis characterized by sudden, severe attacks of burning joint pain, usually the big toe, because of the deposition of uric acid crystals in the joint. The attacks can be recurrent and last a few days to many weeks until there is pain relief. Gout is a disease of high levels of uric acid in the blood and is ten times more common in men than women. Colchicine is effective to prevent and to treat acute attacks.
FMF is a hereditary inflammatory disorder caused by mutations in the MEFV gene that causes episodes of fever, pain and swelling in the abdomen (peritonitis), lungs (pleuritis), heart (pericarditis) and joints (arthritis) in adults and children. Signs and symptoms of FMF usually begin in childhood with attacks that last for weeks or months. Colchicine is considered the gold standard and the only drug recommended for treating FMF. Colchicine is used at low doses to prevent and treat these FMF patients chronically.
Colchicine has a narrow therapeutic index which means that the doses required to treat the disease and the occurrences of serious safety issues are close. Colchicine has common side effects such as abdominal cramping, nausea and diarrhea that have limited its use. More concerning, however, colchicine has "warning and precautions" in the label for drug-drug interactions and should not be taken in conjunction with other drugs that are P-gycoprotein (P-gp) or strong CYP3A4 inhibitors as this could lead to serious side effects and death. Examples of drugs and other items that could increase the concentration of colchicine in the blood into toxic ranges include certain antibiotics, antidepressants, lipid lowering drugs, tranquilizers, grapefruit juice and antihistamines.
APP-111, and its back up APP-112, are NCEs, small molecules that have high oral bioavailability, and like colchicine, bind to the "colchicine binding site" of tubulin. Unlike colchicine, there should not be drug-drug interactions, as APP-111 does not interact with P-gp or CYP3A4. This may potentially eliminate the possibility of serious and life-threatening side effects when given with other drugs that are P-gp or CYP3A4 inhibitors. APP-111/112 could be used as a potentially safer alternative to colchicine, which remains the mainstay of therapy for both prevention and treatment of gout and FMF.
Development Plan. The Phase 1 APP-111 studies that are planned in 2017 and 2018 in prostate cancer patients will provide the initial pharmacokinetics and safety information that can be used for dosing and safety considerations for filing the IND and conducting the Phase 2 studies for gout expected in 2019.
Market. According to nationally representative data (NHANES), gout is the most common form of inflammatory arthritis in men, with a prevalence of 5.9% in men (6.1 million) and in women the prevalence is 2% (2.2 million). The estimated U.S. annual market for gout therapies is $725 million per IMS. FMF affects primarily people of Mediterranean extraction, mostly Sephardic Jews, Armenians, Arabs and Turks. It is very common in the populations at risk with estimated carrier rates of 1/6 in Armenians, 1/7 in North African Jews and 1/13 in Iraqi Jews. FMF affects less than 200,000 patients in the U.S. population and could be eligible for orphan drug status.
11
Consumer Health Product
PREBOOST® (benzocaine 4% wipes) for the prevention of premature ejaculation
Scientific Overview. Premature ejaculation is the most common sexual dysfunction and even more frequent than erectile dysfunction based on epidemiological studies. Premature ejaculation is a self-reported diagnosis. Men with premature ejaculation desire treatment; however, most are reluctant and unlikely to request treatment out of embarrassment. Discrepancies also exist between the man and his partner's reports of the man's ejaculatory behavior as women have been found to report premature ejaculation affecting their relationship more often than their male partner.
There are no FDA-approved prescription products for premature ejaculation. Off-label use of antidepressants and PDE-5 inhibitors have been used with limited success because of inconsistent efficacy and unacceptable side effects. Psychological counseling and behavioral therapy are also used with mixed results. Of the consumer health products, only the topical anesthetics have efficacy and are administered as sprays and gels. The main drawbacks of these products are inconsistent dosing leading to too much anesthetic and transference of the anesthetics to partner. PREBOOST® is compliant with the FDA monograph and is approved for sale in the United States. PREBOOST® is the only individually packaged medicated wipe that contains a weak desensitizing agent (benzocaine 4%). The advantages are: (i) convenient proprietary individually wrapped wipes so it is discreet and easier to carry; (ii) the correct dose is delivered each time; (iii) the medicine is applied topically and dries quickly which prevents the potential for transference to partner; and (iv) benzocaine at 4% is a weak anesthetic that only temporarily desensitizes, but does not completely numb the penis.
Development Plan. PREBOOST® is approved in the United States. The Company has completed an interim analysis of a Phase 4, randomized, double blind placebo controlled study to evaluate the efficacy, safety, and tolerability of PREBOOST® in 21 subjects with premature ejaculation. The scientific abstract that describes the full interim analysis results has been submitted to a major urological medical conference. The independent clinical study was conducted by Jed Kaminetsky, M.D., Medical Director at Manhattan Medical Research, Clinical Assistant Professor of Urology at New York University Medical Center, and practicing urologist with University Urology Associates; Michael Yang, Clinical Research Coordinator at Manhattan Medical Research and University Urology Associates; Michael Perelman. M.D., Clinical Professor Emeritus of Psychology in Psychiatry at Weill Cornell Medical College; and, Ridwan Shabsigh, M.D., Professor of Urology at Weill Cornell Medical College, and President of the International Society of Men’s Health.
The top line results of the interim analysis from 21 men show:
|
· |
After two months, men treated with PREBOOST® had significant improvement in their ability to control ejaculation, with a mean increase in duration of almost four minutes, which was significantly greater than men on placebo. After treatment, 80% of men were no longer considered to have PE; |
|
· |
Men treated with PREBOOST® reported a statistically significant better sense of ejaculation control, confidence, satisfaction, sexual pleasure, length of intercourse and reduced frustration; |
|
· |
PREBOOST® was well tolerated and no transference was reported; and |
|
· |
The interim study results met the primary endpoint of change in average intravaginal ejaculatory latency time (IELT) at two months, and secondary outcomes including change in questionnaire assessments, such as global rating of distress, medication assessment, and Index of Premature Ejaculation (IPE). |
Therefore, the interim analysis of the results of the study showed that PREBOOST® prolonged time to ejaculation, supporting the clinical validity of PREBOOST® for the prevention of premature ejaculation. The Company plans to launch the product in the United States by the end of 2016.
Market. Premature ejaculation is the most prevalent sexual disorder affecting one in four men and is more common than erectile dysfunction. The estimated prevalence is 50 million men in United States and 60 million men in Europe. There are no approved drugs for premature ejaculation and the OTC agents currently available are not optimal or effective. Total worldwide market for premature ejaculation drugs and consumer health care products is estimated to be greater than $500 million. The Company plans to increase sales by having a sampling program targeting urologists, seeking co-promotion opportunities with a marketing partner, introducing the product through additional internet outlets including Walmart, CVS, Walgreens and other OTC distribution outlets, optimizing its internet ecommerce capabilities and digital marketing via www.preboost.com as well as through out-licensing opportunities for markets outside the United States.
FC2 for Global Public Health Sector, as a Consumer Health Product, and as a Medical Device by Prescription.
FC2 for dual protection against unintended pregnancy and STIs
Product. FC2 is the only currently available female-controlled product approved for market by FDA and cleared by the WHO for purchase by U.N. agencies that provides dual protection against unintended pregnancy and sexually transmitted infections STIs, including HIV/AIDS and the Zika virus. FC2 was approved for market by FDA as a Class III medical device in 2009.
12
FC2 has basically the same physical design, specifications, safety, and efficacy profile as FC1, the Company's first generation Female Condom. Manufactured from a nitrile polymer formulation that is exclusive to the Company, FC2 is produced more economically than FC1, which was made from a more costly raw material, polyurethane. FC2 consists of a soft, loose fitting sheath and two rings: an external ring of rolled nitrile and a loose internal ring made of flexible polyurethane. FC2’s soft sheath lines the vagina, preventing skin-to-skin contact during intercourse. Its external ring remains outside the vagina, partially covering the external genitalia. The internal ring is used for insertion and helps keep the device in place during use.
FC2’s primary raw material, a nitrile polymer, offers a number of benefits over natural rubber latex, the raw material most commonly used in male condoms. FC2’s nitrile polymer is stronger than latex, reducing the probability that the female condom sheath will tear during use. Unlike latex, FC2’s nitrile polymer quickly transfers heat. FC2 can warm to body temperature immediately upon insertion, which may enhance the user’s sensation and pleasure. Unlike the male condom, FC2 may be inserted in advance of arousal, eliminating disruption during sexual intimacy. FC2 is also an alternative to latex sensitive users who are unable to use male condoms without irritation. For example, 7 percent to 20 percent of the individuals with significant exposure to latex rubber (i.e., health care workers) experience such irritation. To the Company's knowledge, there is no reported allergy to the nitrile polymer. FC2 is pre-lubricated, disposable, and recommended for use during a single sex act. FC2 is not reusable. In the global public health and consumer health products sectors, FC2 is referred to as the FC2 Female Condom, whereas for the U.S. prescription market, the device is referred to as the Female Disposable Contraceptive Device (FC2).
Global Public Health Sector Market. FC2’s primary use is for disease prevention and family planning, and the global public health sector has been the main market for FC2. Within the public health sector, various organizations supply critical products such as FC2, at no cost or low cost, to those who need but cannot afford to buy such products for themselves.
The Company has a relatively small customer base for FC2, with a limited number of customers who generally purchase in large quantities. Over the past few years, significant customers have included large global agencies, such as the United Nations Population Fund (UNFPA) and the United States Agency for International Development (USAID), Sekunjalo Investments Corporation (PTY) Ltd (Sekunjalo), the Company’s distributor in the Republic of South Africa (RSA), and the Brazil Ministry of Health either through UNFPA or Semina Indústria e Comércio Ltda (Semina), the Company’s distributor in Brazil. Other customers include ministries of health or other governmental agencies, which either purchase directly or via in-country distributors, and non-governmental organizations (NGOs).
FC2 has been distributed in 144 countries. A significant number of countries with the highest demand potential are in the developing world. The incidence of HIV/AIDS, other STIs, and unwanted pregnancy in these countries represents a remarkable potential for significant sales of a product that benefits some of the world’s most underprivileged people. However, conditions in these countries can be volatile and result in unpredictable delays in program development, tender applications, and processing orders.
The global public health sector market for male condoms is estimated to be greater than 8-10 billion units annually. The private sector market for male condoms is estimated at 10-15 billion units annually. The combined global male condom market (public and private sector) is estimated at a value of $4.5 billion annually. The female condom market represents a very small portion of the total global condom market. Yet 50 percent of individuals living with HIV/AIDS are women. As a result a number of independent women’s groups are advocating for increased investment in and distribution of female condoms on a gender equality basis.
Consumer Health Market. The Company has distribution agreements and other arrangements with commercial partners which market as a consumer health product through distributors and retailers in 16 countries, including the United States, Brazil, Spain, France, and the United Kingdom. These agreements are generally exclusive for a single country. Under these agreements, the Company sells FC2 to the distributor partners, who market and distribute the product to consumers in the established territory. An online store for direct-to-consumer purchases, ShopFemaleHealth.com, was launched in March 2015. Additionally, FC2 may now be purchased online through various ecommerce websites, including (but not limited to): Amazon.com, Walgreens.com, CVS.com, Drugstore.com, Kmart.com, Walmart.com and MyQuestStore.com. The Company believes that increased online purchasing of condoms represents an opportunity for the promotion of FC2 to consumers. It is estimated 33 percent of male condoms are purchased online. The Company believes the promotion of FC2 to consumers will be complementary to public sector marketing by increasing awareness of FC2.
13
U.S. Prescription Market. Recent changes in the U.S. market may represent an opportunity for the promotion and expansion of FC2 as a female disposable contraceptive device to protect against STIs and unwanted pregnancies. FC2 is the only device approved by the FDA (Class III device) for this use. As FC2 is nonhormonal, it is a viable alternative for many U.S. women who have reported dissatisfaction with the side effects of hormonal birth control. Moreover, there are unique groups of women such as breast cancer survivors who desire contraception and cannot take hormonal birth control because of this underlying condition. FC2 already has market access as it is currently reimbursable by prescription under the Affordable Care Act such that Medicaid, Medicare, and private health insurance plans are mandated to fully reimburse female birth control products including FC2. FC2 was registered and now has a UPC code to support reimbursement. The Company is implementing a plan to obtain appropriate pricing and market access. In addition to obtaining prescriptions by health care providers in clinics and doctors’ offices, there is an opportunity to obtain an electronic prescription for birth control products, including FC2, with and in certain cases without a physician visit in certain states. We recently hired a Vice President of Marketing with a pharmaceutical background to build the necessary infrastructure to accept prescriptions, organize product fulfillment from independent and central mail order pharmacies, and have payer reimbursement. Marketing and educational programs, both traditional and by digital and social media, are being developed and implemented to target health care providers (physicians, nurse practitioners, and physician assistants), pharmacies, and women to coordinate awareness and access to FC2 as a female disposable contraceptive device that is fully reimbursable.
Government Regulation
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical products and medical devices. These agencies and other federal, state, and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, recordkeeping, tracking, approval, import, export, advertising, and promotion of our products.
FDA Regulation of Female Condoms. Female condoms as a group were classified by the FDA as a Class III medical device in 1989. Class III medical devices are deemed by the FDA to carry potential risks with use which must be tested prior to FDA market approval, referred to as Premarket Approval (PMA), for sale in the U.S. As FC2 is a Class III medical device, prior to selling FC2 in the U.S., the Company was required to submit a PMA application containing technical information on the use of FC2, such as pre-clinical and clinical safety and efficacy studies, which was gathered together in a required format and content. The FC2 PMA was approved for market by the FDA as a Class III medical device in March 2009.
Pursuant to section 515(a)(3) of the Safe Medical Amendments Act of 1990 (the SMA Act), the FDA may temporarily suspend approval and initiate withdrawal of the PMA if the FDA finds that FC2 is unsafe or ineffective, or on the basis of new information with respect to the device, which, when evaluated together with information available at the time of approval, indicates a lack of reasonable assurance that the device is safe or effective under the conditions of use prescribed, recommended, or suggested in the labeling. Failure to comply with the conditions of FDA market approval invalidates the approval order. Commercial distribution of a device that is not in compliance with these conditions is a violation of the SMA Act. As an FDA market approved medical device, the facilities in which FC2 is produced and tested are subject to periodic FDA inspection to ensure compliance with current Good Manufacturing Processes. The Company’s most recent FDA inspection of its U.K. and Malaysian facilities was completed in September 2010. The Chicago office was inspected by the FDA in October 2016 for activities related to being a registered agent.
The FDA’s market approval order for FC2 includes conditions that relate to product labeling, including information on the package itself and instructions for use called a “package insert” which accompanies each product. The Company believes it is in compliance with the FDA market approval order.
FDA Regulation of Pharmaceutical Products. The process required by the FDA before pharmaceutical product candidates may be marketed in the United States generally involves the following:
|
· |
nonclinical laboratory and animal tests, including some that must be conducted in accordance with Good Laboratory Practices; |
|
· |
submission of an IND, which must become effective before clinical trials may begin; |
|
· |
adequate and well‑controlled human clinical trials to establish the safety and efficacy of the proposed drug candidate for its intended use; |
|
· |
pre-approval inspection of manufacturing facilities and selected clinical investigators for their compliance with Good Manufacturing Practices (cGMP) and Good Clinical Practices (cGCP); and |
|
· |
FDA approval of an NDA to permit commercial marketing for particular indications for use. |
14
The testing and approval process requires substantial time, effort, and financial resources. Prior to commencing the first clinical trial with a product candidate, we must submit an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30‑day time period, raises safety concerns or questions about the conduct of the clinical trial by imposing a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Submission of an IND may not result in FDA authorization to commence a clinical trial. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development. Further, an independent institutional review board (IRB) for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial commences at that center. Regulatory authorities or an IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Some studies also include a data safety monitoring board (DSMB), which receives special access to unblinded data during the clinical trial and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy.
In general, for purposes of NDA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
|
· |
Phase 1—Studies are initially conducted to test the product candidate for safety, dosage tolerance, absorption, metabolism, distribution, and excretion in healthy volunteers or patients. |
|
· |
Phase 2—Studies are conducted with groups of patients with a specified disease or condition to provide enough data to evaluate the preliminary efficacy, optimal dosages and dosing schedule, and expanded evidence of safety. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
|
· |
Phase 3—These clinical trials are undertaken in larger patient populations to further evaluate dosage, to provide statistically significant evidence of clinical efficacy, and to further test for safety in an expanded patient population at multiple clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. These trials may be done globally to support global registrations. |
The FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so‑called Phase 4 studies may be made a condition to be satisfied after approval. The results of Phase 4 studies can confirm the effectiveness of a product candidate and can provide important safety information.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product candidate, as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
505(b)(2) Approval Process. Section 505(b)(2) of the Food, Drug and Cosmetic Act (FDCA), which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act, provides an accelerated regulatory pathway to FDA approval for new or improved formulations or new uses of previously approved drug products. Specifically, Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely upon the FDA's findings of safety and effectiveness for an approved product that acts as the Reference Listed Drug (RLD). The FDA may also require 505(b)(2) applicants to perform additional studies or measurements to support the change from the RLD. The FDA may then approve the new product candidate for all or some of the labeled indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
We expect our Tamsulosin DRS, MSS-722 and APP-944 product candidates to qualify for the 505(b)(2) regulatory pathway because they are or will be based on already approved active pharmaceutical ingredients rather than new chemical entities, and formulations that has been through Phase 1 studies. On August 12, 2016, FDA cleared Tamsulosin DRS for the accelerated 505(b)(2) regulatory approval pathway and agreed with our plans to conduct a single bioequivalence study to support the filing of an NDA. On December 6, 2016, based on positive regulatory recommendations by the BRUD Advisory Committee, we plan to file an investigational new drug application (IND) and possibly initiate a Phase 2 clinical study in 2017.
15
Orange Book Listing. In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents whose claims cover the applicant’s product. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Any applicant who files a 505(b)(2) NDA referencing a drug listed in the Orange Book must certify to the FDA that (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is not sought until after patent expiration; or (iv) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product. This last certification is known as a Paragraph IV certification. If the competitor has provided a Paragraph IV certification to the FDA, the competitor must also send notice of the Paragraph IV certification to the holder of the NDA for the RLD and the patent owner once the application has been accepted for filing by the FDA. The NDA holder or patent owner may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification prevents the FDA from approving the application until the earlier of 30 months from the date of the lawsuit, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the applicant. The applicant may also elect to submit a “Section VIII” statement certifying that its proposed label does not contain, or carves out, any language regarding the patented method‑of‑use rather than certify to a listed method‑of‑use patent.
NDA Submission and Review by the FDA. The results of product development, nonclinical studies, and clinical trials are submitted to the FDA as part of an NDA. The submission of an NDA requires payment of a substantial user fee to the FDA. The FDA may convene an advisory committee to provide clinical insight on application review questions. The FDA reviews applications to determine, among other things, whether a product is safe and effective for its intended use and whether the manufacturing controls are adequate to assure and preserve the product’s identity, strength, quality, and purity. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Once the NDA submission has been accepted for filing, which occurs, if at all, within 60 days after submission of the NDA, the FDA’s goal for a non‑priority review of a 505(b)(2) NDA is ten months to complete the review process for the application and respond to the applicant, which can take the form of either a Complete Response Letter or Approval. The review process is often significantly extended by FDA requests for additional information, studies, or clarification. The FDA may delay or refuse approval of an NDA if applicable regulatory criteria are not satisfied, require additional testing or information, and/or require post‑marketing testing and surveillance to monitor safety or efficacy of a product. FDA approval of any NDA submitted by us will be at a time the FDA chooses. Also, if regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. In addition, the FDA may require Phase 4 post‑marketing studies to monitor the effect of approved products, and may limit further marketing of the product based on the results of these post‑marketing studies.
Post‑Approval Requirements. Any products manufactured or distributed by us pursuant to FDA approvals will be subject to continuing regulation by the FDA, including recordkeeping requirements and reporting of adverse experiences. Drug and biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, which impose certain procedural and documentation requirements upon us and our third‑party manufacturers. We cannot be certain that we or our present or future suppliers will be able to comply with the cGMP regulations and other FDA regulatory requirements. If our present or future suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a product from distribution, or withdraw approval of the NDA.
The FDA closely regulates the marketing and promotion of drugs. A company can make only those claims relating to safety and efficacy, purity, and potency that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off‑label uses are common across medical specialties. Physicians may believe that such off‑label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off‑label use.
The recently enacted Drug Supply Chain Security Act imposes new obligations on manufacturers of pharmaceutical products related to product tracking and tracing. Among the requirements of this new legislation, manufacturers will be required to provide certain information regarding the drug products to individuals and entities to which product ownership is transferred, label drug product with a product identifier, and keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers will eventually be required to be done electronically. Manufacturers will also be required to verify that purchasers of the manufacturers’ products are appropriately licensed. Further, under this new legislation, manufacturers will have drug product investigation, quarantine, disposition, and notification responsibilities related to counterfeit, diverted, stolen, and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
16
Other Healthcare Regulations. Our business activities, including but not limited to, research, sales, promotion, distribution, medical education, and other activities will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to the FDA, including potentially the Department of Justice, the Department of Health and Human Services and its various divisions, including the Centers for Medicare and Medicaid Services, and state and local governments. Our business activities must comply with numerous healthcare laws, including but not limited to, the federal Anti‑Kickback Statute, the False Claims Act, the Veterans Health Care Act, and similar state laws.
The federal Anti‑Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting, or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for purchasing, leasing, ordering, or arranging for the purchase, lease, or order of any item or service reimbursable under Medicare, Medicaid, or other federal healthcare programs. The term remuneration has been interpreted broadly to include anything of value. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. The exceptions and safe harbors are drawn narrowly and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchasing, or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti‑Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case‑by‑case basis based on a cumulative review of all of its facts and circumstances.
The federal False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to, or approval by, the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government.
We and our business activities are subject to the civil monetary penalties statute, which imposes penalties against any person or entity who, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
Additionally, the federal Physician Payments Sunshine Act within the Patient Protection and Affordable Care Act (PPACA), and its implementing regulations, require certain manufacturers of drugs and medical devices for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program (with certain exceptions) to report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. The Health Insurance Portability and Accountability Act of 1996 (HIPAA), as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to business associates—independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
The Veterans Health Care Act of 1992 requires manufacturers of “covered drugs” to offer those drugs for sale to certain federal agencies, including but not limited to, the Department of Veterans Affairs, on the Federal Supply Schedule, which requires compliance with applicable federal procurement laws.
Depending on the circumstances, failure to comply with these laws can result in penalties, including criminal, civil, and/or administrative criminal penalties, damages, fines, disgorgement, exclusion of products from reimbursement under government programs, “qui tam” actions brought by individual whistleblowers in the name of the government, refusal to allow us to enter into supply contracts, including government contracts, reputational harm, diminished profits, and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our business.
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals designed to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
17
The Foreign Corrupt Practices Act. The Foreign Corrupt Practices Act (FCPA) prohibits any U.S. individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party, or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
Foreign and Other Regulation. In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products to the extent we choose to develop or sell any products outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from country to country.
FC2 received the CE Mark which allows it to be marketed throughout the European Union. FC2 has also been approved by regulatory authorities in Brazil, India, Canada, and other jurisdictions.
The Company’s facility may also be subject to inspection by UNFPA, USAID, International Organization for Standardization (ISO), and country specific ministries of health.
Intellectual Property
Our success will significantly depend upon our ability to obtain and maintain patent and other intellectual property and proprietary protection for our products. In addition to patents, we rely upon unpatented trade secrets, know‑how, and continuing technological innovation to develop and maintain our competitive position.
As of December 1, 2016, we owned or held exclusive rights to 9 issued U.S. patents, 6 pending U.S. patent applications and additional patents and patent applications in other jurisdictions. These include a provisional patent application relating to our Tamsulosin DRS product that is subject to deferred payment obligations and patents and patent applications relating to our APP-111 and APP-112 drug candidates that we license from a third party. Additional information regarding our patent portfolio is provided below.
PREBOOST® Patent Application. PREBOOST®, medicated individual wipes which is a male genital desensitizing drug product that helps in the prevention of premature ejaculation, is covered by a pending U.S. patent application.
MSS-722 Patent. MSS-722, an oral agent we are developing for the treatment of male infertility, is covered by an issued U.S. patent that expires in 2021. We also own additional foreign patents and patent applications covering MSS-722.
Tamsulosin DRS Patent Application. We own a provisional patent application with respect to Tamsulosin DRS. APP acquired those patent rights pursuant to a purchase agreement that provides for significant continuing installment and milestone payment obligations. In addition, APP granted a security interest in the purchased assets to the seller to secure APP's present and future payment and performance obligations under the purchase agreement. Accordingly, there will be significant payments that APP will be required to make in the future to the seller of the Tamsulosin DRS assets and the failure to make such payments may result in APP losing its rights to such intellectual property. If APP fails to retain such rights, we would not be able to commercialize any products relating to Tamsulosin DRS.
APP-944 Patent Application. We own one U.S. provisional patent application related to the development of an oral drug for the treatment of hot flashes in men on prostate cancer therapies.
APP-111/APP-112 License. We hold an exclusive license to 5 issued U.S. patents, 2 pending U.S. patent applications and 63 patents and patent applications in other jurisdictions relating to our APP-111 and APP-112 drug candidates. This license contains provisions requiring up-front, milestone and royalty payments to the licensor. If we fail to comply with these obligations or other obligations to the licensor, the licensor might have the right to terminate the license, in which event we would not be able to commercialize these drug candidates.
FC2 Patents. FC2 patents have been issued by the United States, Europe, Canada, Australia, South Africa, the People’s Republic of China, Japan, Mexico, Brazil, India and the African Regional Intellectual Property Organization (ARIPO), which includes Botswana, Gambia, Ghana, Kenya, Lesotho, Malawi, Mozambique, Namibia, Sierra Leone, Sudan, Swaziland, Tanzania, Uganda, Zambia, and Zimbabwe. Further, the European patent for FC2 has been validated in the following countries: Austria, Belgium, Bulgaria, Switzerland, Republic of Cyprus, Czech Republic, Germany, Denmark, Estonia, Spain, Finland, France, United Kingdom, Greece, Hungary, Ireland, Italy, Luxembourg, Monaco, Netherlands, Portugal, Romania, Sweden, Slovenia, Slovakia, and Turkey. The
18
patents cover the key aspects of FC2, including its overall design and manufacturing process. The patents have expiration dates in 2023 and 2024.
Trademarks. The Company has a registration for the trademarks “FC2 Female Condom” and "PREBOOST" in the United States and has filed applications in the U.S. for the trademarks "Veru Healthcare," "Veru Biopharma," "Veru Pharmaceuticals" and "Veru Pharma." The Company has filed applications or secured registrations in 40 countries or jurisdictions around the world to protect the various names and symbols used in marketing its Female Condoms.
Significant Customers
Because FC2 provides dual protection against both STIs, including HIV/AIDS, and unintended pregnancy, it is an integral part of both HIV/AIDS prevention and family planning programs throughout the world. These programs are typically supplied by global public health sector buyers who purchase products for distribution, at low cost or no cost, to those who need but cannot afford to buy such products themselves. Within the global public health sector are large global agencies, such as UNFPA, USAID, DFID (the U.K.’s Department for International Development), and PSI (Population Services International), other social marketing groups, various government health agencies, and NGOs. The Company’s most significant customers are either global public health sector agencies, country specific ministries of health, or those who facilitate their purchases and/or distribution.
The Company's three largest customers in fiscal 2016 were the Brazil Ministry of Health (through Semina), UNFPA, and USAID. Semina accounted for 27 percent of unit sales in fiscal 2016, 47 percent of unit sales in fiscal 2015, and less than 10 percent of unit sales in fiscal 2014. UNFPA accounted for 25 percent of unit sales in fiscal 2016, 18 percent of unit sales in fiscal 2015, and 40 percent of unit sales in fiscal 2014. USAID accounted for 24 percent of unit sales in fiscal 2016, 16 percent of unit sales in fiscal 2015, and 17 percent of unit sales in fiscal 2014. Sekunjalo and Azinor International Lda, a customer in Angola (Azinor), accounted for 13 and 11 percent of unit sales, respectively, in fiscal 2014. No other single customer accounted for more than 10 percent of unit sales in fiscal 2016, 2015, or 2014. The Company considers its most significant customers to be UNFPA, USAID, Sekunjalo, and the Brazil Ministry of Health (either through UNFPA or Semina).
Employees
As of December 9, 2016, the Company had 155 full-time employees, including 12 located in the U.S., 14 in the U.K., 127 in Malaysia, and 2 in other countries to implement training and programs, and 1 part-time employee located in the U.S. None of the Company’s employees are represented by a labor union. The Company believes that its employee relations are good. In Malaysia, a significant proportion of direct labor is supplied by a contracted work force.
Environmental Regulation
The Company believes there are no material issues or material costs associated with the Company's compliance with environmental laws related to the manufacture and distribution of FC2. The Company has not incurred environmental expenses in fiscal 2016, 2015, or 2014, nor does it anticipate environmental expenses in the foreseeable future.
Raw Materials
The principal raw material used to produce FC2 is a nitrile polymer. While general nitrile formulations are available from a number of suppliers, the Company has chosen to work closely with the technical market leader in synthetic polymers to develop a grade ideally suited to the bio-compatibility and functional needs of a female condom. As a result, the Company relies on supply for its principal raw material from one supplier that could produce the raw material from multiple supply points within its organization.
Manufacturing
The Company leases production space in Selangor D.E., Malaysia for the production of FC2, which currently has manufacturing capacity of approximately 100 million units annually. In fiscal 2014 the Company added additional space, resulting in a total of 45,800 sq. ft. in the Company’s Malaysia facility, comprised of production and warehouse space and which provides sufficient space to add manufacturing capacity of up to an additional 100 million units annually. The Company will consider manufacturing in other locations as the demand for FC2 develops.
The Company expects to rely on third-party contract manufacturers and other third parties to produce, package and store sufficient quantities of any future drug candidates. The Company has entered into an agreement with a third-party contract manufacturer to produce its PREBOOST® medicated individual wipes for managing premature ejaculation.
19
Competition
FC2 participates in the same market as male condoms; however, it is not seen as directly competing with male condoms. Rather, studies show that providing FC2 is additive in terms of prevention and choice. Male condoms cost less and have brand names that are more widely recognized than FC2. In addition, male condoms are generally manufactured and marketed by companies with significantly greater financial resources than the Company.
Other parties have developed and marketed female condoms. None of these female condoms marketed or under development by other parties have secured FDA market approval. FDA market approval is required to sell female condoms in the U.S. USAID, a U.S. government funded agency, is required to procure from the FDA product approval for market; however there can be exceptions. Outside of the U.S., the Company has experienced increasing competition and pricing pressures for FC2. In addition to FC2, three female condoms have successfully completed the WHO prequalification process and been cleared by UNFPA for purchase by U.N. agencies: the Cupid female condom (which was prequalified by WHO in July 2012 and cleared by UNFPA thereafter), the Velvet female condom marketed by Hindustan Latex Limited (which was prequalified by WHO and cleared by UNFPA in March 2016) and the female condom marketed by PATH (which was prequalified by WHO and cleared by UNFPA in March 2016). The female condom marketed by Hindustan Latex Limited, which is the Company’s former exclusive distributor in India, is substantially similar in design to FC2, except it is made of latex. FC2 has also been competing with other female condoms in markets that do not require either FDA market approval or WHO prequalification. Reflecting increased competition, Cupid received part of the last two South African tenders. Increasing competition in FC2’s markets has, and will likely continue to, put pressure on pricing for FC2 and may also adversely affect sales of FC2. Some customers, particularly in the global public sector, prioritize price over other features where FC2 may have an advantage. It is also possible that other female condoms may receive FDA market approval or complete the WHO prequalification process, which would increase competition from other female condoms in FC2’s markets.
The pharmaceutical industry is highly competitive, and is characterized by extensive research efforts and rapid technological progress. The success of our pharmaceutical products will depend on our ability to acquire, develop and commercialize products and our ability to establish and maintain markets for any products for which we receive marketing approval. Potential competitors in North America, Europe and elsewhere include major pharmaceutical companies, specialty pharmaceutical companies and biotechnology firms, universities and other research institutions and government agencies. Many of the competitors with respect to our pharmaceutical products under development have substantially greater research and development and regulatory capabilities and experience, and substantially greater management, manufacturing, distribution, marketing and financial resources, than we will.
All drugs currently used to treat BPH symptoms are tablets or capsules. These drugs include those that shrink the prostate, like 5 alpha reductase inhibitors which include PROSCAR (finasteride) from Merck & Co., Inc. and AVODART (dutasteride) from GlaxoSmithKline. The other major class of drugs treat BPH by relaxing the smooth muscles of the prostate and bladder neck and include alpha blockers like FLOMAX® (tamsulosin HCI) from Boehringer Ingelheim Pharmaceuticals, HYTRIN (terazocin), UROXATRAL (alfuzosin), CARDURA (doxazosin), and RAPAFLO (silodosin) from Allergan as well as Phosphodiesterase 5 (PDE5) inhibitors like CIALIS (tadalafil) from Eli Lilly. One class of drugs combines a drug that shrinks and another that relaxes the prostate called JALYN (dutasteride/tamsulosin combination) from GlaxoSmithKline.
There are drugs that have been approved for the treatment of male infertility for the indication of hypogonadotropic hypogonadism. These drugs are only available by injection which include: Gonal-F® (Follitropin Alfa) which is recombinant DNA human follicle stimulating hormone by EMD Serono, Inc., Novarel® (chorionic gonadotropin for injection USP) which is human chorionic gonadotropin by Ferring, Inc. and Pregnyl® (chorionic gonadotropin for injection USP) which is human chorionic gonadotropin by Merck & Co., Inc. There are no FDA-approved oral therapies for male infertility. CLOMID (clomiphene citrate) 50mg tablets are being used off-label using various doses and dosing schedules for idiopathic infertile men. CLOMID and generics are FDA-approved as 50 mg dose for the treatment of ovulatory dysfunction in women desiring pregnancy. CLOMID and generics of CLOMID contain a mixture of two geometric isomers cis-clomiphene (zuclomiphene) and trans-clomiphene (enclomiphene), and contain between 30-50% of the cis-clomiphene isomer.
Although there are no FDA-approved drugs for the treatment of hot flashes in men who have advanced prostate cancer as a side effect of prostate cancer hormone therapies, there are several drugs being used off-label including estrogens and selective serotonin reuptake inhibitor antidepressants including EFFEXOR (venlafaxine) and anticonvulsants like NEURONTIN (gabapentin) which could be competitive with our APP-944 product candidate for the treatment of hot flashes in men who have advanced prostate cancer as a side effect of prostate cancer hormone therapies.
20
APP-111 is a first-in-class oral chemotherapy that targets the colchicine binding site of tubulin and will be initially developed for prostate, breast and ovarian cancers. All currently available tubulin targeting chemotherapies are given IV, not orally, and include: Vinca Alkaloids such as VELBAN® (vinblastine), ONCOVIN® (vincristine) and NAVELBINE® (vinorelbine). These chemotherapies are primarily used for hematologic malignancies (leukemia, lymphoma, myeloma, sarcoma), and some neuroblastoma, thyroid cancer and nonsmall cell cancer of the lung. Taxanes such as TAXOL® (paclitaxel), TAXOTERE® (docetaxel) and JEVTANA® (cabazitaxel) are primarily used for solid tumors such as breast, ovarian, endometrial, cervical, lung, head and neck, esophageal, bladder, gastric and prostate. TAXOTERE® (docetaxel) and JEVTANA® (cabazitaxel) are indicated for advanced metastatic prostate cancer, are given IV and bind to the taxane site of tubulin.
The main therapeutic products that are competitive with PREBOOST™ include lidocaine and other anesthetic creams, gels and sprays. Off-label use of selective serotonin reuptake inhibitor antidepressants like PAXIL® (paroxetine) have also been used off-label to prevent premature ejaculation.
Backlog
Unfilled product orders totaled $2,829,535 at December 9, 2016 and $7,386,526 at November 27, 2015. Unfilled orders materially fluctuate from quarter-to-quarter, and the amount at December 9, 2016 includes orders with requested delivery dates later in fiscal 2017. The Company expects current unfilled orders to be filled during fiscal 2017.
Available Information
The Company maintains a corporate website for investors at www.veruhealthcare.com/investor and it makes available, free of charge, through this website its annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports that the Company files with or furnishes to the Securities and Exchange Commission (SEC), as soon as reasonably practicable after it electronically files such material with, or furnishes it to, the SEC. Information on the Company's website is not part of this report.
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, together with all of the other information included in this Annual Report and our other SEC filings, in considering our business and prospects. The risks described below are not the only risks we face. Additional risks that we do not yet know of or that we currently think are immaterial may also impair our business operations. If any of the events or circumstances described in the following risks occur, our business, financial condition, results of operations or prospects could be materially adversely affected. In such cases, the trading price of our common stock could decline.
Risks Related to Our Business
We may be unable to realize the benefits anticipated by the APP Merger or it may take longer than anticipated for the combined company to achieve those benefits.
Our realization of the benefits anticipated as a result of the APP Merger will depend in part on the integration of APP's business with that of FHC under a new management team led by our new President and Chief Executive Officer, Mitchell S. Steiner, M.D. However, we may not be able to effectively operate and integrate APP's business and FHC's business. The dedication of management resources to this integration could detract attention from the day-to-day business of FHC and APP, and FHC cannot assure shareholders that there will not be substantial costs associated with the transition process or other negative consequences as a result of these integration efforts. These effects, including, but not limited to, incurring unexpected costs or delays in connection with integration of the two businesses, or the failure of APP's business to perform as expected, could harm our results of operations.
Additional financing will be needed to support our development activities.
We expect to incur significant expenditures over the next several years to support our preclinical and clinical development activities, particularly with respect to clinical trials for our MSS-722, APP-944, APP-111 and APP-112 drug candidates and the bioequivalence study for Tamsulosin DRS. This will require us to obtain additional capital for our business, and additional capital may not be available on terms acceptable to us.
If we are unable to obtain needed financing on acceptable terms, we may not be able to implement our business plan, which could have a material adverse effect on our business, financial condition, results of operations and prospects. If we raise additional funds through the sale of equity, convertible debt or other equity-linked securities, our shareholders' ownership will be diluted. We may issue securities that have rights, preferences and privileges senior to our common stock.
21
If adequate funds cannot be raised, we may be required to:
|
· |
delay, reduce the scope of or eliminate one or more of our development programs; or |
|
· |
relinquish, license or otherwise dispose of rights to technologies, drug candidates or products that we would otherwise seek to develop or commercialize itself at an earlier stage or on terms that are less favorable than might otherwise be available. |
Our future capital requirements will depend upon a number of factors, including:
|
· |
the size, complexity, results and timing of our development programs; |
|
· |
the cost to obtain sufficient supply of the compounds necessary for our drug candidates at a reasonable cost; |
|
· |
the time and cost involved in obtaining regulatory approvals; |
|
· |
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims; |
|
· |
the costs involved in manufacturing and commercializing our drug candidates; and |
|
· |
competing technological and market developments. |
These factors could result in variations from currently projected operating and liquidity requirements.
Our business may be affected by contracting risks with government and other international health agencies.
Our customers for FC2 have primarily been large international agencies and government health agencies which purchase and distribute FC2 for use in family planning and HIV/AIDS prevention programs. Sales to such agencies may be subject to government contracting risks, including the appropriations process and funding priorities, potential bureaucratic delays in awarding contracts under governmental tenders, process errors, politics or other pressures, and the risk that contracts may be subject to cancellation, delay, or restructuring. A governmental tender award indicates acceptance of the bidder’s price rather than an order or guarantee of the purchase of any minimum number of units. Many governmental tenders are stated to be “up to” the maximum number of units, which gives the applicable government agency discretion to purchase less than the full maximum tender amount. As a result, government agencies may order and purchase fewer units than the full maximum tender amount and there are no guarantees as to the timing or amount of actual orders or shipments under government tenders. Orders received may vary from the amount of the tender award based on a number of factors, including vendor supply capacity, quality inspections, and changes in demand. These contracting risks may cause significant quarter-to-quarter variations in our operating results and could adversely affect our net revenues and profitability. Budget issues, spending cuts, and global health spending priorities affecting government health agencies may also adversely affect demand for FC2 and our net revenues.
We will experience intense competition.
We are engaged in the marketing and development of products in industries, including the pharmaceutical industry, that are highly competitive. The pharmaceutical industry is also characterized by extensive research and rapid technological progress. Potential competitors with respect to our drug candidates in North America, Europe and elsewhere include major pharmaceutical companies, specialty pharmaceutical companies and biotechnology firms, universities and other research institutions and government agencies. Many of our competitors have substantially greater research and development and regulatory capabilities and experience, and substantially greater management, manufacturing, distribution, marketing and financial resources, than we have. We may be unable to compete successfully against current and future competitors, and competitive pressures could have a negative effect on our net revenues and profit margins.
Other parties have developed and marketed female condoms, although only three such products have WHO pre-clearance and none of these female condoms have been approved for market by the FDA. FDA market approval is required to sell female condoms in the U.S., and WHO pre-clearance is required to sell female condoms to U.N. agencies. FC2 has also been competing with other female condoms in markets that do not require either FDA market approval or WHO prequalification. We have experienced increasing competition in the global public sector, and competitors received part of the last two South African tenders. Increasing competition in FC2’s markets may put pressure on pricing for FC2 or adversely affect sales of FC2, and some customers, particularly in the global public sector, may prioritize price over other features where FC2 may have an advantage. It is also possible that other companies will develop a female condom, and such companies could have greater financial resources and customer contacts than us. In addition, other contraceptive methods may compete with FC2 for funding and attention in the global public sector.
An inability to identify or complete future acquisitions could adversely affect our future growth.
We intend to pursue acquisitions of new products, technologies, and/or businesses that enable us to leverage our competitive strengths. While we continue to evaluate potential acquisitions, we may not be able to identify and successfully negotiate suitable acquisitions, obtain financing for future acquisitions on satisfactory terms, obtain regulatory approval for acquisitions where required, or otherwise complete acquisitions in the future. An inability to identify or complete future acquisitions could limit our future growth.
22
We may experience difficulties in integrating strategic acquisitions.
The integration of acquired companies and their operations into our operations involves a number of risks, including:
|
· |
the acquired business may experience losses that could adversely affect our profitability; |
|
· |
unanticipated costs relating to the integration of acquired businesses may increase our expenses; |
|
· |
possible failure to accomplish the strategic objectives for an acquisition; |
|
· |
the loss of key personnel of the acquired business; |
|
· |
difficulties in achieving planned cost-savings and synergies may increase our expenses or decrease our net revenues; |
|
· |
diversion of management’s attention could impair their ability to effectively manage our business operations; |
|
· |
the acquired business may require significant expenditures for product development or regulatory approvals; |
|
· |
the acquired business may lack adequate internal controls or have other issues with its financial systems; |
|
· |
there may be regulatory compliance or other issues relating to the business practices of an acquired business; |
|
· |
we may record goodwill and nonamortizable intangible assets that are subject to impairment testing on a regular basis and potential impairment charges and we may also incur amortization expenses related to intangible assets; and |
|
· |
unanticipated management or operational problems or liabilities may adversely affect our profitability and financial condition. |
Additionally, we may borrow funds or issue equity to finance strategic acquisitions. Debt leverage resulting from future acquisitions could adversely affect our operating margins and limit our ability to capitalize on future business opportunities. Such borrowings may also be subject to fluctuations in interest rates. Equity issuances may dilute our existing shareholders and adversely affect the market price of our shares.
We depend on four major customers for a significant portion of our net revenues.
The Company's four largest customers currently are UNFPA, USAID, Sekunjalo and Semina. UNFPA accounted for 25 percent of unit sales in fiscal 2016, 18 percent of unit sales in fiscal 2015, and 40 percent of unit sales in fiscal 2014. USAID accounted for 24 percent of unit sales in fiscal 2016, 16 percent of unit sales in fiscal 2015, and 17 percent of unit sales in fiscal 2014. Sekunjalo accounted for less than 10 percent of unit sales in fiscal 2016 and 2015 and 13 percent of unit sales in fiscal 2014. Semina accounted for 27 percent of unit sales in fiscal 2016, 47 percent of unit sales in fiscal 2015, and less than 10 percent of unit sales in fiscal 2014. An adverse change in our relationship with our largest customers could have a material adverse effect on our net revenues and profitability. In addition, we may have a concentration of accounts receivable with one or more of our largest customers, and a delay in payment by a large customer could have a material adverse effect on our cash flows and liquidity.
Since we sell product in foreign markets, we are subject to international business risks that could adversely affect our operating results.
Our international operations subject us to risks, including:
|
· |
economic and political instability; |
|
· |
changes in international regulatory requirements, import duties, or export restrictions, including limitations on the repatriation of earnings; |
|
· |
difficulties in staffing and managing foreign operations; |
|
· |
complications in complying with trade and foreign tax laws; |
|
· |
price controls and other restrictions on foreign currency; and |
|
· |
difficulties in our ability to enforce legal rights and remedies. |
Any of these risks might disrupt the supply of our products, increase our expenses or decrease our net revenues. The cost of compliance with trade and foreign tax laws increases our expenses, and actual or alleged violations of such laws could result in enforcement actions or financial penalties that could result in substantial costs.
Increases in the cost of raw materials, labor, and other costs used to manufacture FC2 could increase our cost of sales and reduce our gross margins.
We may experience increased costs of raw materials, including the nitrile polymer used in FC2, and increased labor costs. We may not be able to pass along such cost increases to our customers. As a result, an increase in the cost of raw materials, labor or other costs associated with manufacturing FC2 could increase our cost of sales and reduce our gross margins.
23
Currency exchange rate fluctuations could increase our expenses.
Because we manufacture FC2 in a leased facility located in Malaysia, a portion of our operating costs are denominated in a foreign currency. While a material portion of our future sales of FC2 are likely to be in foreign markets, all sales of FC2 are denominated in U.S. dollars. Manufacturing costs are subject to normal currency risks associated with fluctuations in the exchange rate of the Malaysian ringgit (MYR) relative to the U.S. dollar. Historically, we have not hedged our foreign currency risk.
We rely on a single facility to manufacture FC2, which subjects us to the risk of supply disruptions.
We manufacture FC2 in a single leased facility located in Malaysia. Difficulties encountered by this facility, such as fire, accident, natural disaster, or an outbreak of a contagious disease could halt or disrupt production at the facility, delay the completion of orders, or cause the cancellation of orders. Any of these risks could increase our expenses or reduce our net revenues.
Uncertainty and adverse changes in the general economic conditions may negatively affect our business.
If general economic conditions in the U.S. and other global markets in which we operate decline, or if consumers fear that economic conditions will decline, consumers may reduce expenditures for products such as our products. Adverse changes may occur as a result of adverse global or regional economic conditions, fluctuating oil prices, declining consumer confidence, unemployment, fluctuations in stock markets, contraction of credit availability, or other factors affecting economic conditions generally. These changes may negatively affect the sales of our products, increase the cost, and decrease the availability of financing, or increase costs associated with producing and distributing our products. In addition, a substantial portion of the sales of FC2 are made in the public market to government agencies, including USAID and other government agencies around the world. Worsening economic conditions as well as budget deficits and austerity measures may cause pressures on government budgets and result in a reduction in purchases of FC2 by governmental agencies. Sales of FC2 fluctuate, which causes our operating results to vary from quarter-to-quarter.
Sales of FC2 fluctuate based upon demand from our commercial partners and the public sector and the nature of government procurement processes. Historically, our net revenues and profitability have varied from quarter–to-quarter due to such buying patterns. Quarterly variations in operating results may cause us to fail to meet our earnings guidance or market expectations for our operating results and may tend to depress our stock price during such quarters.
Material adverse or unforeseen legal judgments, fines, penalties, or settlements could have an adverse impact on our profits and cash flows.
We may, from time to time, become a party to legal proceedings incidental to our business, including, but not limited to, alleged claims relating to product liability, environmental compliance, patent infringement, commercial disputes, and employment matters. The current and future use of our drug candidates by us and potential collaborators in clinical trials, and the sale of any approved products in the future, may expose us to product liability claims. We will face an inherent risk of product liability claims as a result of the clinical testing of our drug candidates, and will face an even greater risk if we obtain FDA approval and commercialize our drug candidates in the U.S. or other additional jurisdictions or if we engage in the clinical testing of proposed new products or commercialize any additional products. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourself against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our existing products or drug candidates, if approved. Regardless of the merits or eventual outcome, product liability claims may result in any of the following:
|
· |
the inability to commercialize our drug candidates; |
|
· |
difficulty recruiting subjects for clinical trials or withdrawal of these subjects before a trial is completed; |
|
· |
labeling, marketing, or promotional restrictions; |
|
· |
product recalls or withdrawals; |
|
· |
decreased demand for our products or products that we may develop in the future; |
|
· |
loss of revenue; |
|
· |
injury to reputation; |
|
· |
initiation of investigations by regulators; |
|
· |
costs to defend the related litigation; |
|
· |
substantial monetary awards to trial participants or patients; and |
|
· |
a decline in the value of our shares. |
24
Litigation could require us to record reserves or make payments which could adversely affect our profits and cash flows. Even the successful defense of legal proceedings may cause us to incur substantial legal costs, may divert management's attention and resources away from our business, may prevent us or our partners from achieving or maintaining market acceptance of the affected product and may substantially increase the costs of commercializing our future products and impair the ability to generate revenues from the commercialization of these products either by us or by our strategic alliance partners.
We currently maintain limited general commercial liability insurance coverage. However, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or for liabilities in excess of our insurance limits, our assets may not be sufficient to cover such claims and our business operations could be impaired.
In connection with the APP Merger, two putative class action and derivative lawsuits were filed against us and our directors alleging breach of fiduciary duty and/or wasting of corporate assets. These lawsuits are currently in a very early stage. Any unfavorable outcomes in these lawsuits, resulting in the payment of damages or affecting our transaction with APP, could have a material adverse effect on our business and prospects and could reduce our profitability. In addition, addressing these lawsuits will likely divert management's attention and resources from our business.
Risks Related to the Regulation and Commercialization of Our Products and Drug Candidates
The Company has no experience in obtaining regulatory approval for a drug.
Although our President and Chief Executive Officer has experience in obtaining regulatory approval for a drug under development, the Company has never obtained regulatory approval for, or commercialized, a drug. It is possible that the FDA may refuse to accept any or all of our planned NDAs for substantive review or may conclude, after review of our data, that our applications are insufficient to obtain regulatory approval of any of our drug candidates. The FDA may also require that we conduct additional clinical or manufacturing validation studies, which may be costly and time-consuming, and submit that data before it will reconsider our applications. Depending on the extent of these or any other FDA required studies, approval of any NDA that we submit may be significantly delayed, possibly for years, or may require us to expend more resources than we have available or can secure. Any delay or inability in obtaining regulatory approvals would delay or prevent us from commercializing our drug candidates, generating revenue from these proposed products and achieving and sustaining profitability. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA to approve any NDA we submit. If any of these outcomes occur, we may be forced to abandon our planned NDAs for one or more of our drug candidates, which would materially adversely affect our business.
Clinical trials involve a lengthy and expensive process with an uncertain outcome and results of earlier studies and trials may not be predictive of future trial results. Clinical trials are expensive, can take many years to complete and have highly uncertain outcomes. Failure can occur at any time during the clinical trial process as a result of inadequate performance of a drug, inadequate adherence by patients or investigators to clinical trial protocols or other factors. New drugs in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through earlier clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials as a result of a lack of efficacy or adverse safety profiles, despite promising results in earlier trials. Our future clinical trials may not be successful or may be more expensive or time-consuming than we currently expect. If clinical trials for any of our drug candidates fail to demonstrate safety or efficacy to the satisfaction of the FDA, the FDA will not approve that drug and we would not be able to commercialize it, which will have a material adverse effect on our business, financial condition, results of operations and prospects.
We could experience delays in our planned clinical trials.
We may experience delays in clinical trials that will be required to be conducted for our drug candidates. Our planned clinical trials might not begin on time; may be interrupted, delayed, suspended, or terminated once commenced; might need to be redesigned; might not enroll a sufficient number of patients; or might not be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including the following:
|
· |
delays in obtaining regulatory approval to commence a trial; |
|
· |
imposition of a clinical hold following an inspection of our clinical trial operations or trial sites by the FDA or other regulatory authorities; |
|
· |
imposition of a clinical hold because of safety or efficacy concerns by the FDA, a DSMB, a clinical trial site's IRB or us; |
|
· |
delays in reaching agreement on acceptable terms with prospective contract research organizations (CROs) and clinical trial sites; |
|
· |
delays in obtaining required IRB approval at each site; |
|
· |
delays in identifying, recruiting and training suitable clinical investigators; |
|
· |
delays in recruiting suitable patients to participate in a trial; |
25
|
· |
delays in having patients complete participation in a trial or return for post-treatment follow-up; |
|
· |
clinical sites dropping out of a trial to the detriment of enrollment; |
|
· |
time required to add new sites; |
|
· |
delays in obtaining sufficient supplies of clinical trial materials, including suitable active pharmaceutical ingredients; or |
|
· |
delays resulting from negative or equivocal findings of DSMB for a trial. |
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials, and clinicians' and patients' perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. Any of these delays in completing our clinical trials could increase our costs, slow down our product development and approval process and jeopardize our ability to commence product sales and generate revenue as to the affected drug candidate.
Our clinical trials may be suspended or discontinued.
Before we can obtain regulatory approval for the commercial sale of our MSS-722, APP-944, APP-111 and APP-112 drug candidates, we are required to complete preclinical development with respect to such product candidates and/or extensive clinical trials in humans to demonstrate its safety and efficacy. To date, regulatory approval has not been obtained for any of our drug candidates.
Unfavorable results from preclinical studies or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated. In addition, we may report top-line data from time to time, which is based on a preliminary analysis of key efficacy and safety data. Such top-line data may be subject to change following a more comprehensive review of the data related to the applicable clinical trial. If we delay or abandon our development efforts related to our MSS-722, APP-944, APP-111 or APP-112 drug candidates, or any other potential future drug candidate fails to demonstrate sufficient safety and efficacy in any clinical trial, we would experience potentially significant delays in, or be required to abandon, development of that drug candidate. If we delay or abandon our development efforts related to any of the MSS-722, APP-944, APP-111 or APP-112 drug candidates, or any other potential future drug candidate, our business, financial condition, results of operations and prospects may be materially adversely affected.
Our clinical trials may be suspended or terminated at any time for a number of reasons. A clinical trial may be suspended or terminated by us, our collaborators, the FDA or other regulatory authorities because of a failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, presentation of unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using the investigational drug, changes in governmental regulations or administrative actions, lack of adequate funding to continue the clinical trial or negative or equivocal findings of the DSMB or the IRB for a clinical trial. An IRB may also suspend or terminate our clinical trials for failure to protect patient safety or patient rights. We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants. In addition, regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe the clinical trials are not being conducted in accordance with applicable regulatory requirements or present an unacceptable safety risk to participants. If we elect or are forced to suspend or terminate any clinical trial of any proposed product that we develop, the commercial prospects of such proposed product will be harmed and our ability to generate product revenue from any of these proposed products will be delayed or eliminated. Any of these occurrences may materially harm our business, financial condition, results of operations and prospects.
We may be subject to risks relating to collaboration with third parties.
As part of our business strategy, we may enter into collaboration arrangements with strategic partners to develop and commercialize our drug candidates. For our collaboration efforts to be successful, we must identify partners whose competencies complement our competencies. We may be unsuccessful in entering into collaboration agreements with acceptable partners or negotiating favorable terms in these agreements. Also, we may be unsuccessful in integrating the resources and capabilities of these collaborators with our own. In addition, we may face a disadvantage in seeking to enter into or negotiating collaborations with potential partners because other potential collaborators may have greater management and financial resources than we do. Our collaborators may prove difficult to work with or less skilled than originally expected. If we are unsuccessful in our collaborative efforts, our ability to develop and market drug candidates could be severely limited.
26
We intend to rely on CROs to conduct our research and development activities.
We will not have the resources to independently conduct research and development activities. Therefore, we intend to rely on CROs and universities to conduct research and development activities for our drug candidates and for the execution of our clinical studies. Although we will control only certain aspects of our CROs' activities, we will be responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We cannot be sure that the CROs will conduct the research properly or in a timely manner, or that the results will be reproducible. We and our CROs are required to comply with the FDA's cGCPs, which are regulations and guidelines enforced by the FDA for all of our drug products in clinical development. The FDA enforces these cGCPs through periodic inspections of trial sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable or invalid and the FDA may require us to perform additional clinical trials before approving our drug candidates. In addition, to evaluate the safety and effectiveness compared to placebo of our drug candidates to a statistically significant degree, our clinical trials will require an adequately large number of test subjects. Any clinical trial that a CRO conducts abroad on our behalf is subject to similar regulation. Accordingly, if our CROs fail to comply with these regulations or recruit a sufficient number of patients, we may be required to repeat clinical trials, which would delay the regulatory approval process.
In addition, we will not employ the personnel of our CROs, and, except for remedies available to us under our agreements with such organizations, we cannot control whether or not they will devote sufficient time and resources to our research and development and our clinical studies. Our CROs may also have relationships with other commercial entities, including one or more of our competitors, for which they may also be conducting clinical studies or other drug development activities, which could impede their ability to devote appropriate time to our clinical programs. If our CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised because of the failure to adhere to our clinical protocols or regulatory requirements, or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our drug candidates that we seeks to develop. As a result, our financial results and the commercial prospects for our drug candidates that we seek to develop would be harmed, our costs could increase and our ability to generate revenue from such drug candidates could be delayed or ended.
If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. Switching or entering into new relationships with CROs involves substantial cost and requires extensive management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially affect our ability to meet our desired clinical development timelines and can increase our costs significantly. We may encounter challenges or delays in entering into or maintaining these relationships, and any such delays or challenges may have a material adverse impact on our business, financial condition, results of operations and prospects.
We expect to rely on third party manufacturers for our drug candidates.
For the foreseeable future, we expect to rely on third-party manufacturers and other third parties to produce, package and store sufficient quantities of any future drug candidates for use in our clinical trials. These drug candidates are complicated and expensive to manufacture. If our future third-party manufacturers fail to deliver our drug candidates for clinical use on a timely basis, with sufficient quality, and at commercially reasonable prices, we may be required to delay or suspend clinical trials or otherwise discontinue development and production of our drug candidates. While we may be able to identify replacement third-party manufacturers or develop our own manufacturing capabilities for these drug candidates, this process would likely cause a delay in the availability of our drug candidates and an increase in costs. In addition, third-party manufacturers may have a limited number of facilities in which our drug candidates can be produced, and any interruption of the operation of those facilities due to events such as equipment malfunction or failure or damage to the facility by natural disasters could result in the cancellation of shipments, loss of product in the manufacturing process or a shortfall in available drug candidates.
27
In addition, regulatory requirements could pose barriers to the manufacture of our drug candidates. Third-party manufacturers are required to comply with the FDA's cGMPs. As a result, the facilities used by any of future manufacturers of our drug candidates must be approved by the FDA. Holders of NDAs, or other forms of FDA approvals or clearances, or those distributing a regulated product under their own name, are responsible for manufacturing even though that manufacturing is conducted by a third-party contract manufacturing organization (CMO). Our third-party manufacturers will be required to produce our drug candidates under FDA cGMPs in order to meet acceptable standards for our clinical trials. Our third-party manufacturers may not perform their obligations under their agreements with us or may discontinue their business before the time required by us to gain approval for or commercialize our drug candidates. In addition, our manufacturers will be subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. Failure by any of our manufacturers to comply with applicable cGMPs could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspensions or withdrawals of approvals, operating restrictions, interruptions in supply, recalls, withdrawals, issuance of safety alerts and criminal prosecutions, any of which could have a material adverse impact on our business, financial condition, results of operations and prospects. Finally, we also could experience manufacturing delays if our CMOs give greater priority to the supply of other products over our products or otherwise do not satisfactorily perform according to the terms of their agreements with us.
If any supplier of the product for our drug candidates experiences any significant difficulties in its respective manufacturing processes, does not comply with the terms of the agreement between us or does not devote sufficient time, energy and care to providing our manufacturing needs, we could experience significant interruptions in the supply of our drug candidates, which could impair our ability to supply our drug candidates at the levels required for our clinical trials and commercialization and prevent or delay their successful development and commercialization.
Changes in law, including as a result of the recent presidential and congressional elections, could have a negative impact on the approval of our drug candidates.
The FDA has established regulations, guidelines and policies to govern the drug development and approval process, as have foreign regulatory authorities. Any change in regulatory requirements resulting from the adoption of new legislation, regulations or policies may require us to amend existing clinical trial protocols or add new clinical trials to comply with these changes. Such amendments to existing protocols or clinical trial applications or the need for new ones, may significantly and adversely affect the cost, timing and completion of the clinical trials for our drug candidates. In addition, the FDA's policies may change and additional government regulations may be issued that could prevent, limit or delay regulatory approval of our drug candidates, or impose more stringent product labeling and post-marketing testing and other requirements. The recent presidential and congressional elections in the U.S. could result in significant changes in, and uncertainty with respect to, legislation, regulation and government policy that could significantly impact our business and the health care industry. While it is not possible to predict whether and when any such changes will occur, specific proposals discussed during and after the election that could have a material impact on us include, but are not limited to, the repeal of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 and enactment of the 21st Century Cures Act. If we are slow or unable to adapt to any such changes, our business, prospects and ability to achieve or sustain profitability would be adversely affected.
We may fail to commercialize our drug candidates.
We cannot be sure that, if our clinical trials for any of our MSS-722, APP-944, APP-111 or APP-112 drug candidates are successfully completed, we will be able to submit an NDA to the FDA or that any NDA we submit will be approved by the FDA in a timely manner, if at all. We also cannot be sure that, if our bioequivalence study for Tamsulosin DRS is successfully completed, any NDA we submit will be approved by the FDA in a timely manner, if at all. After completing clinical trials for a drug candidate in humans, a drug dossier is prepared and submitted to the FDA as an NDA, and includes all preclinical studies and clinical trial data relevant to the safety and effectiveness of the product at the suggested dose and duration of use for the proposed indication as well as manufacturing information, in order to allow the FDA to review such drug dossier and to consider a drug candidate for approval for commercialization in the United States. If we are unable to submit an NDA with respect to any of the MSS-722, APP- 944, APP-111, APP-112 or Tamsulosin DRS, or if any NDA we submit is not approved by the FDA, we will be unable to commercialize that product. The FDA can and does reject NDAs and require additional clinical trials, even when drug candidates achieve favorable results in Phase 3 clinical trials. If we fail to commercialize any of the MSS-722, APP-944, APP-111 or APP-112 drug candidates, or Tamsulosin DRS, our business, financial condition, results of operations and prospects may be materially adversely affected and our reputation in the industry and in the investment community would likely be damaged.
28
We are subject to extensive and costly governmental regulation.
Our products, including FC2 and our drug candidates, are subject to extensive and rigorous domestic government regulation, including regulation by the FDA, the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services, including its Office of Inspector General, the U.S. Department of Justice, the Departments of Defense and Veterans Affairs, to the extent our products are paid for directly or indirectly by those departments, state and local governments and their respective foreign equivalents. The FDA regulates the research, development, preclinical and clinical testing, manufacture, safety, effectiveness, record keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import and export of pharmaceutical products and medical devices under various regulatory provisions. Any of our products that are tested or marketed abroad are also be subject to extensive regulation by foreign governments, whether or not we have obtained FDA approval for a given product and its uses. Such foreign regulation may be equally or more demanding than corresponding U.S. regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing and selling products. Our failure to comply with these regulations could result in, by way of example, significant fines, criminal and civil liability, product seizures, recalls, withdrawals, withdrawals of approvals and exclusion and debarment from government programs. Any of these actions, including the inability of our products to obtain and maintain regulatory approval, would have a materially adverse effect on our business, financial condition, results of operations and prospects.
We are subject to additional health care regulation and enforcement by the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include the following:
|
· |
the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order, or recommendation of, any good or service for which payment may be made under government health care programs such as the Medicare and Medicaid programs; |
|
· |
the federal False Claims Act that prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other government health care programs that are false or fraudulent; |
|
· |
federal criminal laws that prohibit executing a scheme to defraud any health care benefit program or making false statements relating to health care matters; and |
|
· |
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians for marketing. Some states, such as California, Massachusetts and Vermont, mandate implementation of corporate compliance programs, along with the tracking and reporting of gifts, compensation and other remuneration to physicians.
The scope and enforcement of these laws is uncertain and subject to change in the current environment of health care reform, especially in light of the lack of applicable precedent and regulations. We cannot predict the impact on our business of any changes in these laws. Federal or state regulatory authorities may challenge our current or future activities under these laws. Any such challenge could have a material adverse effect on our reputation, business, results of operations and financial condition. Any state or federal regulatory review of us, regardless of the outcome, would be costly and time-consuming.
We could experience misconduct by our employees.
We will be exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with federal and state health care fraud and abuse laws and regulations, to comply with the FCPA, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and prevent employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
29
Coverage and reimbursement may not be available for our products.
Market acceptance and sales for MSS-722, APP-944, APP-111, APP-112 and Tamsulosin DRS, if approved, will depend on coverage and reimbursement policies and may be affected by health care reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which products they will pay for and establish reimbursement levels. We cannot be sure that coverage and reimbursement will be available for our drug candidates, if approved. We also cannot be sure that the amount of reimbursement available, if any, will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our drug candidates.
We may not be able to gain and retain market acceptance for our drug candidates.
Physicians may not prescribe our drug candidates, if approved by the appropriate regulatory authorities for marketing and sale, which would prevent any such drug candidate from generating revenue. Market acceptance of our drug candidates, by physicians, patients and payors, will depend on a number of factors, many of which are beyond our control, including the following:
|
· |
the clinical indications for which our drug candidates are approved, if at all; |
|
· |
acceptance by physicians and payors of each product as safe and effective treatment; |
|
· |
the cost of treatment in relation to alternative treatments; |
|
· |
the relative convenience and ease of administration of our products in the treatment of the symptoms for which they are intended; |
|
· |
the availability and efficacy of competitive drugs; |
|
· |
the effectiveness of our sales force and marketing efforts; |
|
· |
the extent to which the product is approved for inclusion on formularies of hospitals and managed care organizations; |
|
· |
the availability of coverage and adequate reimbursement by third parties, such as insurance companies and other health care payors, or by government health care programs, including Medicare and Medicaid; |
|
· |
limitations or warnings contained in a product's FDA-approved labeling; and |
|
· |
prevalence and severity of adverse side effects. |
Even if the medical community accepts that our drug candidates are safe and efficacious for their approved indications, physicians may not immediately be receptive to the use or may be slow to adopt such products as an accepted treatment for the symptoms for which they are intended. We cannot be sure that any labeling approved by the FDA will permit us to promote our products as being superior to competing products. If our drug candidates, if approved, do not achieve an adequate level of acceptance by physicians and payors, we may not generate sufficient or any revenue from these products. In addition, our efforts to educate the medical community and third-party payors on the benefits of our products may require significant resources and may never be successful.
In addition, even if our drug candidates achieve market acceptance, we may not be able to maintain that market acceptance over time if:
|
· |
new products or technologies are introduced that are more favorably received than our products, are more cost effective or render our products obsolete; |
|
· |
unforeseen complications arise with respect to use of our products; or |
|
· |
sufficient third-party insurance coverage or reimbursement does not remain available. |
In many foreign markets, including the countries in the European Union, pricing of pharmaceutical products is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing controls. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our profitability.
Third parties may obtain FDA regulatory exclusivity to our detriment.
We plan to seek to obtain market exclusivity for our drug candidates and any other drug candidates we develop in the future. To the extent that patent protection is not available or has expired, FDA marketing exclusivity may be the only available form of exclusivity available for these proposed products. Marketing exclusivity can delay the submission or the approval of certain marketing applications. Potentially competitive products may also be seeking marketing exclusivity and may be in various stages of development, including some more advanced than our products. We cannot predict with certainty the timing of FDA approval or whether FDA approval will be granted, nor can we predict with certainty the timing of FDA approval for competing products or whether such approval will be granted. It is possible that competing products may obtain FDA approval with marketing exclusivity before we do, which could delay our ability to submit a marketing application or obtain necessary regulatory approvals, result in lost market opportunities with respect to our drug candidates and materially adversely affect our business, financial condition and results of operations.
30
Risks Relating to Our Intellectual Property
We may be unable to protect the proprietary nature of the intellectual property covering our products.
Our commercial success will depend in part on our ability to obtain patents, as well as our ability to maintain adequate protection of other intellectual property for our drug candidates and other products. If we do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and profitability. The patent positions of pharmaceutical products are highly uncertain. The legal principles applicable to patents are in transition due to changing court precedent and legislative action and we cannot be certain that the historical legal standards surrounding questions of validity will continue to be applied or that current defenses relating to issued patents in these fields will be sufficient in the future. Changes in patent laws in the United States, such as the America Invents Act of 2011, may affect the scope, strength and enforceability of our patent rights or the nature of proceedings that may be brought by us related to our patent rights. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States and we may encounter significant problems in protecting our proprietary rights in these countries. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies are covered by valid and enforceable patents or are effectively maintained as trade secrets.
These risks include the possibility of the following:
|
· |
the patent applications that we have filed may fail to result in issued patents in the United States or in foreign countries; |
|
· |
patents issued or licensed to us or our partners may be challenged or discovered to have been issued on the basis of insufficient, incomplete or incorrect information, and thus held to be invalid or unenforceable; |
|
· |
the scope of any patent protection may be too narrow to exclude competitors from developing or designing around these patents; |
|
· |
the scope of any patent protection may be too narrow to exclude competitors from developing or designing around these patents; |
|
· |
we or our licensors were not the first to make the inventions covered by each issued patents and pending patent applications; |
|
· |
we or our licensors were not the first inventors to file patent applications for these technologies in the United States or were not the first to file patent applications directed to these technologies abroad; |
|
· |
we may fail to comply with procedural, documentary, fee payment and other similar provisions during the patent application process, which can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights; |
|
· |
future drug candidates may not be patentable; |
|
· |
others will claim rights or ownership with regard to patents and other proprietary rights that we hold or license; |
|
· |
delays in development, testing, clinical trials and regulatory review may reduce the period of time during which we could market our drug candidates under patent protection; and |
|
· |
we may fail to timely apply for patents on our technologies or products. |
While we will apply for patents covering our technologies and products, as we deem appropriate, many third parties may already have filed patent applications or have received patents in our areas of product development. These entities' applications, patents and other intellectual property rights may conflict with patent applications to which we have rights and could prevent us from obtaining patents or could call into question the validity of any of our patents, if issued, or could otherwise adversely affect our ability to develop, manufacture, commercialize or market our products. In addition, if third parties file patent applications in the technologies that also claim technology to which we have rights, we may have to participate in interference, derivation or other proceedings with the U.S. Patent and Trademark Office (USPTO), or foreign patent regulatory authorities to determine our rights in the technologies, which may be time-consuming and expensive. Moreover, issued patents may be challenged in the courts or in post-grant proceedings at the USPTO, or in similar proceedings in foreign countries. These proceedings may result in loss of patent claims or adverse changes to the scope of the claims.
If we or our licensors or strategic partners fail to obtain and maintain patent protection for our products, or our proprietary technologies and their uses, companies may be dissuaded from collaborating with us. In such event, our ability to commercialize our drug candidates or future drug candidates, if approved, may be threatened, we could lose our competitive advantage and the competition we face could increase, all of which could adversely affect our business, financial condition, results of operations and prospects.
In addition, mechanisms exist in much of the world permitting some form of challenge by generic drug marketers to patents prior to, or immediately following, the expiration of any regulatory exclusivity, and generic companies are increasingly employing aggressive strategies, such as "at risk" launches to challenge relevant patent rights.
31
Our business also may rely on unpatented proprietary technology, know-how, and trade secrets. If the confidentiality of this intellectual property is breached, it could adversely impact our business.
We are dependent in part on some license relationships.
We have acquired by license technology relating to our APP-111 and APP-112 drug candidates, and might enter into additional licenses in the future. Licenses to which we are a party contain, and we expect that any future licenses will contain, provisions requiring up-front, milestone and royalty payments to licensors. If we fail to comply with these obligations or other obligations to a licensor, that licensor might have the right to terminate the license on relatively short notice, in which event we would not be able to commercialize the drug candidates that were covered by the license. Also, the milestone and other payments associated with these licenses will make it less profitable for us to develop our drug candidates.
We have continuing obligations under our purchase agreement to acquire the patent rights with respect to Tamsulosin DRS and granted a lien on those assets in connection with such acquisition.
In addition to an upfront payment that we made in connection with the acquisition of the patent rights associated with Tamsulosin DRS, there are significant installment payments and milestone payments that are required to be made pursuant to the terms of the purchase agreement. In addition, we granted a security interest in the purchased assets to the seller to secure our present and future payment and performance obligations under the purchase agreement. Accordingly, there will be significant payments that we will be required to make in the future to the seller of the Tamsulosin DRS assets and the failure to make such payments may result in us losing our rights to such intellectual property. If we fail to retain such rights, we would not be able to commercialize any products relating to Tamsulosin DRS. In such event, our business, results of operations, financial condition and prospects would be materially adversely affected.
We may face claims that our intellectual property infringes on the intellectual property rights of third parties.
Our success depends, in part, on not infringing the patents and proprietary rights of other parties and not breaching any license, collaboration or other agreements we enter into with regard to our technologies and products. Numerous United States and foreign issued patents and pending patent applications owned by others also exist in the therapeutic areas in, and for the therapeutic targets for, which we intend to develop drugs. Patent applications are confidential when filed and remain confidential until publication, approximately 18 months after initial filing, while some patent applications remain unpublished until issuance. As such, there may be other third-party patents and pending applications of which we will be unaware with claims directed towards composition of matter, formulations, methods of manufacture, or methods for treatment related to the use or manufacture of our products or drug candidates. Therefore, we cannot know with certainty the nature or existence of every third-party patent filing. We cannot be sure that us or our partners will be free to manufacture or market our drug candidates as planned or that us or our licensors' and partners' patents will not be opposed or litigated by third parties. If any third-party patent was held by a court of competent jurisdiction to cover aspects of our materials, formulations, methods of manufacture or methods of treatment related to the use or manufacture of any of our drug candidates, the holders of any such patent may be able to block our ability to develop and commercialize the applicable drug candidate unless we obtained a license or until such patent expires or is finally determined to be held invalid or unenforceable. We may not be able to obtain a license to such patent on favorable terms or at all. Failure to obtain such license may have a material adverse effect on our business.
There is a substantial amount of litigation involving intellectual property in the pharmaceutical industry. If a third party asserts that we infringe its patents or other proprietary rights, we could face a number of risks that could adversely affect our business, financial condition, results of operations and prospects, including the following:
|
· |
infringement and other intellectual property claims, which would be costly and time-consuming to defend, whether or not we are ultimately successful, which in turn could delay the regulatory approval process, consume our capital and divert management's attention from our business; |
|
· |
substantial damages for past infringement, which we may have to pay if a court determines that our products or technologies infringe a competitor's patent or other proprietary rights; |
|
· |
a court prohibiting us from selling or licensing our technologies or future products unless the third party licenses its patents or other proprietary rights to us on commercially reasonable terms, which it is not required to do; |
|
· |
redesigning our products so they do not infringe, which may not be possible or may require substantial monetary expenditures and time. |
We cannot predict whether third parties will assert these claims against us or our strategic partners or against the licensors of technology licensed to us, or whether those claims will harm our business. In addition, the outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. If we or our partners were to face infringement claims or
32
challenges by third parties relating to our drug candidates, an adverse outcome could subject us to significant liabilities to such third parties, and force us or our partners to curtail or cease the development of some or all of our drug candidates, which could adversely affect our business, financial condition, results of operations and prospects.
We must submit patent certifications in connection with the 505(b)(2) FDA regulatory pathway.
We intend to submit NDAs for our MSS-722 and APP-944 drug candidates, assuming that the clinical data justify submission, and for our Tamsulosin DRS product under Section 505(b)(2) of the FDCA, which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the filing of an NDA when at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. To the extent that a Section 505(b)(2) NDA relies on clinical trials conducted for a previously approved drug product or the FDA's prior findings of safety and effectiveness for a previously approved drug product, the Section 505(b)(2) applicant must submit patent certifications in its Section 505(b)(2) NDA with respect to any patents for the approved product on which the application relies that are listed in the FDA's publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Specifically, the applicant must certify for each listed patent that (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is not sought until after patent expiration; or (iv) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product. A certification that the new product will not infringe the previously approved product's listed patent or that such patent is invalid or unenforceable is known as a Paragraph IV certification.
If the Section 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the owner of the referenced NDA for the previously approved product and relevant patent holders within 20 days after the Section 505(b)(2) NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement suit against the Section 505(b)(2) applicant. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification prevents the FDA from approving the application until the earlier of 30 months from the date of the lawsuit, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the applicant. The court also has the ability to shorten or lengthen the 30 month period if either party is found not to be reasonably cooperating in expediting the litigation. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its product only to be subject to significant delay and patent litigation before its product may be commercialized. Alternatively, if the NDA or relevant patent holder does not file a patent infringement lawsuit within the specified 45 day period, the FDA may approve the Section 505(b)(2) application at any time.
If we cannot certify that all of the patents listed in the Orange Book for the approved products referenced in the NDAs for each of our drug candidates have expired, we will be compelled to include a Paragraph IV certification in the NDA for such drug candidate. Our inability to certify that all of the patents listed in the FDA's Orange Book for approved products referenced in the NDAs for each of our drug candidates could have a serious and significant adverse effect on the timing for obtaining approval of our drug candidates.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of our competitors.
As is common in the pharmaceutical industry, we will employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees, or we, have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Such claims may lead to material costs for us, or an inability to protect or use valuable intellectual property rights, which could adversely affect our business, financial condition, results of operations and prospects.
We may need to file lawsuits or take other actions to protect or enforce our intellectual property rights.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. Moreover, we may not have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights, generally.
33
In addition, in an infringement proceeding, a court may decide that one of our patents or one of our licensor's patents is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents, or those of ours licensors, do not cover the technology in question or on other grounds. An adverse result in any litigation or defense proceedings could put one or more of our patents, or those of our licensors, at risk of being invalidated, held unenforceable or interpreted narrowly and could put our patent applications, or those of our licensors, at risk of not issuing. Moreover, we may not be able to prevent, alone or with our licensors, misappropriation of our proprietary rights, particularly in countries in which the laws may not protect those rights as fully as in the United States or in those countries in which we do not file national phase patent applications. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. The occurrence of any of the above could adversely affect our business, financial condition, results of operations and prospects.
We may fail to protect the confidentiality of commercially sensitive information.
We will rely on trade secrets, know-how and continuing technological advancement to develop and maintain our competitive position. To protect this competitive position, we will enter into confidentiality and proprietary information agreements with third parties, including employees, independent contractors, suppliers and collaborators. We cannot, however, ensure that these protective arrangements will be honored by third parties and we may not have adequate remedies if these arrangements are breached. In addition, enforcement of claims that a third party has illegally obtained and is using trade secrets, know-how or technological advancements is expensive, time-consuming and uncertain. Non-U.S. courts are sometimes less willing than U.S. courts to protect this information. Moreover, our trade secrets, know-how and technological advancements may otherwise become known or be independently developed by competitors in a manner providing us with no practical recourse against the competing parties. If any such events were to occur, they could adversely affect our business, financial condition, results of operations and prospects.
Risks Related to Ownership of Our Common Stock
A limited number of shareholders may be able to exercise substantial influence over us.
Assuming the conversion of all of the shares of Series 4 Preferred Stock into FHC Common Stock in accordance with their terms, Mitchell S. Steiner, M.D. will beneficially own approximately 14.5% of the shares of FHC Common Stock outstanding and Harry Fisch, M.D. will beneficially own approximately 14.6% of the shares of FHC Common Stock outstanding. These shareholders may have the ability to exert significant influence over the outcome of shareholder votes, including votes concerning director elections, amendments to our Articles of Incorporation and possible mergers, corporate control contests and other significant corporate transactions.
Charges to earnings resulting from the APP Merger may cause our operating results to suffer.
Under the acquisition method of accounting in accordance with ASC 805, Business Combinations, we will allocate the total purchase price of the APP Merger to APP's net tangible assets and intangible assets based on their respective fair values as of the date of the APP Merger, and we will record the excess of the purchase price over those fair values as goodwill. Management's estimates of the fair value of such assets will be based upon assumptions that they believe to be reasonable but that will be inherently uncertain. The following factors, among others, could result in material charges that would cause our financial results to be negatively impacted:
|
· |
impairment of goodwill; |
|
· |
charges for the amortization of identifiable intangible assets and for stock-based compensation; |
|
· |
accrual of newly identified pre-merger contingent liabilities that are identified subsequent to the finalization of the purchase price allocation; and |
|
· |
charges to income to eliminate certain of FHC's pre-merger activities that duplicate those of APP or to reduce the combined company's cost structure. |
Additional costs may include costs of employee redeployment, relocation and retention, including salary increases or bonuses, accelerated amortization of deferred equity compensation and severance payments, reorganization or closure of facilities, taxes and termination of contracts that provide redundant or conflicting services. Some of these costs may have to be accounted for as expenses that would decrease net income and earnings per share for the periods in which those adjustments are made.
34
If we fail to maintain effective internal control over financial reporting, our ability to produce accurate financial statements or comply with applicable regulations could be impaired.
Pursuant to Section 404 of the Sarbanes-Oxley Act, our management is required annually to deliver a report that assesses the effectiveness of our internal control over financial reporting and our independent registered public accounting firm is required annually to deliver an attestation report on the effectiveness of our internal control over financial reporting. If we are unable to maintain effective internal control over financial reporting or if our independent auditors are unwilling or unable to provide us with an attestation report on the effectiveness of internal control over financial reporting for future periods as required by Section 404 of the Sarbanes-Oxley Act, we may not be able to produce accurate financial statements, and investors may therefore lose confidence in our operating results, our stock price could decline and we may be subject to litigation or regulatory enforcement actions.
There are provisions in our charter documents, Wisconsin law, and our credit agreement that might prevent or delay a change in control of our company.
We are subject to a number of provisions in our charter documents, Wisconsin law, and change of control agreements that may discourage, delay, or prevent a merger or acquisition that a shareholder may consider favorable. These provisions include the following:
|
· |
the authority provided to our Board of Directors in our Amended and Restated Articles of Incorporation to issue preferred stock without further action by our shareholders; |
|
· |
the provision under Wisconsin law that permits shareholders to act by written consent only if such consent is unanimous; |
|
· |
the provision under Wisconsin law that requires for a corporation such as us, that was formed before January 1, 1973, the affirmative vote of the holders of at least two-thirds of the outstanding shares of our voting stock to approve an amendment to our articles of incorporation, a merger submitted to a vote of our shareholders, or a sale of substantially all of our assets; |
|
· |
advance notice procedures for nominations of candidates for election as directors and for shareholder proposals to be considered at shareholders’ meetings; |
|
· |
covenants in our credit agreement restricting mergers, asset sales and similar transactions and a provision in our credit agreement that triggers an event of default upon the acquisition by a person or a group of persons of beneficial ownership of 25% or more of our outstanding common stock; and |
|
· |
the Wisconsin control share acquisition statute and Wisconsin's "fair price" and "business combination" provisions which limit the ability of an acquiring person to engage in certain transactions or to exercise the full voting power of acquired shares under certain circumstances. |
The trading price of our common stock has been volatile, and investors in our common stock may experience substantial losses.
The trading price of our common stock has been volatile and may become volatile again in the future. The trading price of our common stock could decline or fluctuate in response to a variety of factors, including:
|
· |
our failure to meet market expectations for our performance; |
|
· |
the timing of announcements by us or our competitors concerning significant product developments, acquisitions, or financial performance; |
|
· |
fluctuation in our quarterly operating results; |
|
· |
substantial sales of our common stock; |
|
· |
general stock market conditions; or |
|
· |
other economic or external factors. |
You may be unable to sell your stock at or above your purchase price.
If our stock price declines, our common stock may be subject to delisting from the NASDAQ Capital Market.
If the closing bid price of our common stock is less than $1.00 per share for 30 consecutive trading days, we may receive a letter from the staff of The NASDAQ Stock Market LLC stating that our common stock will be delisted unless we are able to regain compliance with the Nasdaq Listing Rule requiring that we maintain a closing bid price for our common stock of at least $1.00 per share. The closing bid price of our common stock was below $1.00 per share for a number of days recently, but not for 30 consecutive trading days. We cannot guarantee that our stock price will continue to trade above $1.00 per share or otherwise meet the NASDAQ listing requirements and therefore our common stock may in the future be subject to delisting. If our common stock is delisted, this would, among other things, substantially impair our ability to raise additional funds and could result in a loss of institutional investor interest and fewer development opportunities for us.
35
Future sales of our common stock may depress our stock price.
Sales of a substantial number of shares of our common stock in the public market, or the perception that these sales might occur may reduce the prevailing market price of our common stock and make it more difficult for you to sell your common stock at a time and price that you deem appropriate. In addition, the former stockholders of APP are entitled to rights with respect to the registration of the shares they received pursuant to the APP Merger under the Securities Act of 1933, as amended (the Securities Act). Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act. Any sales of securities by existing shareholders could have a material adverse effect on the market price of our common stock.
Item 1B. Unresolved Staff Comments
Not Applicable
In May 2016, the Company entered a new lease agreement for approximately 6,600 sq. ft. of office space in Chicago, IL. The Chicago lease expires October 31, 2023, although the Company has a renewal option to extend the term for a period of five years. In November 2016, the Company entered into a new lease for approximately 2,600 sq. ft of office space in Miami, FL. The Miami lease expires on November 1, 2019, although the Company has two renewal options to extend the term for a period of three years each. The Company utilizes warehouse space and sales fulfillment services of an independent public warehouse located in Glendale Heights, IL for storage and distribution of FC2 and an independent public warehouse in Lakewood, New Jersey for storage and distribution of PREBOOST®. In June 2010, the Company entered a new lease agreement for 6,400 square feet of office space located in London, England. The lease expires in June 2020. The Company manufactures and warehouses FC2 within a leased facility with 45,800 sq. ft. of production and warehouse space, in Selangor D.E., Malaysia. The FDA-approved manufacturing process is subject to periodic inspections by the FDA as well as the U.K. based “notified body”, which is responsible for CE and ISO accreditation. The lease currently has an expiration date of September 1, 2016 and is renewable at the option of the Company for an additional three year term. The Company’s Malaysian production capacity is approximately 100 million units annually.
On or about October 21, 2016, an alleged FHC shareholder, Martin Glotzer, filed a purported derivative and class action complaint on behalf of himself and the public shareholders of FHC in the Circuit Court of Cook County, Illinois, captioned Glotzer v. The Female Health Company, et al., Case No. 2016-CH-13815. The lawsuit names as defendants FHC and the seven persons who were members of FHC's board of directors prior to the closing of the APP Merger. The complaint alleges, among other things, that FHC's directors breached their fiduciary duties and wasted corporate assets by continuing to expend FHC's resources in soliciting shareholder votes in connection with the APP Merger. Based on these allegations, the complaint seeks equitable relief, including rescinding the APP Merger, damages on behalf of FHC and costs and expenses of the litigation, including attorneys' fees. FHC believes that this action is without merit and intends to defend its position in this matter vigorously.
On or about November 7, 2016, an alleged FHC shareholder, Brian C. Schartz, filed a purported derivative and class action complaint on behalf of himself and all other similarly situated shareholders of FHC in the Circuit Court of Cook County, Illinois, captioned Schartz v. Parrish, et al., Case No. 2016-CH-14488. The lawsuit names as defendants FHC, the members of FHC's board of directors prior to the closing of the APP Merger and the members of FHC's board of directors after the closing of the APP Merger. The complaint alleges, among other things, that FHC's directors breached their fiduciary duties by consummating the APP Merger in violation of the Wisconsin Business Corporation Law and by causing FHC to disseminate to its shareholders press releases and SEC filings containing materially false and misleading statements. Based on these allegations, the complaint seeks equitable relief, including enjoining the FHC Board from taking action in furtherance of the APP Merger, damages on behalf of FHC and costs and expenses of the litigation, including attorneys' fees. On November 18, 2016, the defendants filed to remove the action to United States District Court for the Northern District of Illinois, where the case is captioned Schartz v. Parrish, et al., Case No. 1:16-cv-10736. FHC believes that this action is without merit and intends to defend its position in this matter vigorously.
Item 4. Mine Safety Disclosures
Not Applicable
36
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Shares of our common stock trade on the NASDAQ Capital Market under the symbol "FHCO". The approximate number of record holders of our common stock at December 9, 2016 was 269. The Company has not paid cash dividends on its common stock since May 2014. The Company intends to retain any earnings for use in operations and, therefore, does not anticipate paying cash dividends for the foreseeable future. The Company's credit facility with BMO Harris Bank N.A., restricts dividends and share repurchases. Information regarding the high and low reported closing prices for our common stock is set forth in the table below.
|
|
|||||||||||
|
QUARTERS |
|||||||||||
|
FIRST |
SECOND |
THIRD |
FOURTH |
||||||||
|
2016 Fiscal Year |
|||||||||||
|
Price per common share – High |
$ |
2.01 |
$ |
2.65 |
$ |
1.90 |
$ |
1.47 | |||
|
Price per common share – Low |
$ |
1.38 |
$ |
1.20 |
$ |
1.25 |
$ |
1.17 | |||
|
2015 Fiscal Year |
|||||||||||
|
Price per common share – High |
$ |
4.59 |
$ |
3.92 |
$ |
3.33 |
$ |
1.78 | |||
|
Price per common share – Low |
$ |
3.32 |
$ |
2.80 |
$ |
1.80 |
$ |
1.32 | |||
37
Performance Graph
The performance graph set forth below shows the value of an investment of $100 on September 30, 2011 in each of The Female Health Company, the NASDAQ Composite Index and NASDAQ Health Care Index. All values assume reinvestment of the pre-tax value of dividends paid by FHC and the companies included in the indices, and are calculated as of September 30 each year. The historical stock price performance of FHC is not necessarily indicative of future stock performance.
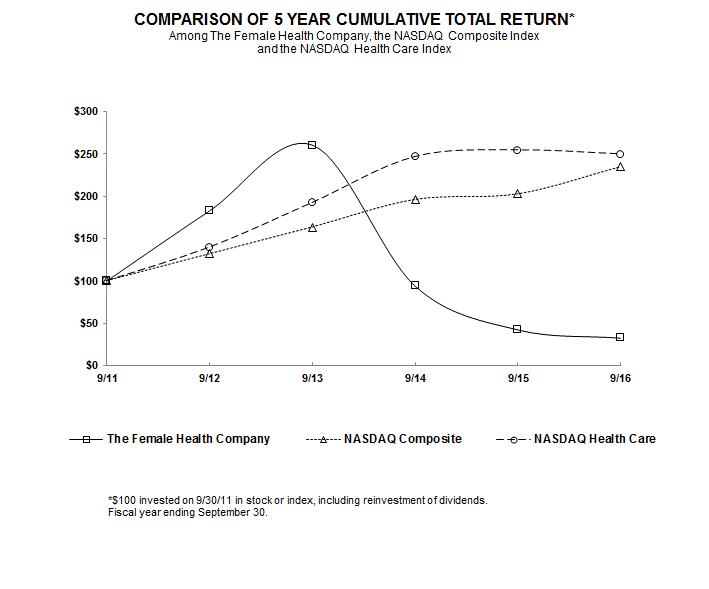
|
|
9/11 |
9/12 |
9/13 |
9/14 |
9/15 |
9/16 |
|
|
||||||
|
The Female Health Company |
100.00 | 182.54 | 260.06 | 94.38 | 42.73 | 32.99 |
|
NASDAQ Composite |
100.00 | 131.89 | 163.47 | 195.96 | 202.60 | 234.66 |
|
NASDAQ Health Care |
100.00 | 139.65 | 192.42 | 246.63 | 254.43 | 249.67 |
38
Item 6. Selected Financial Data
The data set forth below should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Consolidated Financial Statements and Notes thereto appearing in this Annual Report on Form 10-K. The Consolidated Statement of Income Data for the years ended September 30, 2016, 2015, and 2014, and the Consolidated Balance Sheet Data as of September 30, 2016 and 2015, are derived from the Consolidated Financial Statements included elsewhere in this report. The Consolidated Statement of Income Data for the years ended September 30, 2013 and 2012, and the Consolidated Balance Sheet Data as of September 30, 2014, 2013, and 2012, are derived from Consolidated Financial Statements that are not included in this report. The historical results are not necessarily indicative of results to be expected for future periods.
|
|
||||||||||||||
|
|
Year ended September 30, |
|||||||||||||
|
Condensed Consolidated Statement of Income Data: |
2016 |
2015 |
2014 |
2013 |
2012 |
|||||||||
|
(In thousands, except per share data) |
||||||||||||||
|
Net revenues |
$ |
22,127 |
$ |
32,605 |
$ |
24,491 |
$ |
31,457 |
$ |
35,034 | ||||
|
Cost of sales |
8,778 | 13,635 | 11,370 | 13,953 | 14,413 | |||||||||
|
Gross profit |
13,349 | 18,970 | 13,121 | 17,504 | 20,621 | |||||||||
|
Operating expenses |
10,330 | 12,352 | 9,197 | 7,714 | 9,681 | |||||||||
|
Operating income |
3,019 | 6,618 | 3,924 | 9,790 | 10,940 | |||||||||
|
Non-operating income (expense) |
(205) | 69 | 33 | 144 | (148) | |||||||||
|
Income before income taxes |
2,814 | 6,687 | 3,957 | 9,934 | 10,792 | |||||||||
|
Income tax expense (benefit) |
2,469 | 2,341 | 1,524 | (4,409) | (4,507) | |||||||||
|
Net income |
$ |
345 |
$ |
4,346 |
$ |
2,433 |
$ |
14,343 |
$ |
15,299 | ||||
|
Net income per basic common share outstanding |
$ |
0.01 |
$ |
0.15 |
$ |
0.09 |
$ |
0.51 |
$ |
0.55 | ||||
|
Basic weighted average common shares outstanding |
28,666 | 28,532 | 28,523 | 28,377 | 27,694 | |||||||||
|
Net income per diluted common share outstanding |
$ |
0.01 |
$ |
0.15 |
$ |
0.08 |
$ |
0.50 |
$ |
0.53 | ||||
|
Diluted weighted average common shares outstanding |
28,927 | 28,834 | 28,865 | 28,726 | 28,933 | |||||||||
|
Cash dividends declared per share |
$ |
— |
$ |
— |
$ |
0.21 |
$ |
0.26 |
$ |
0.22 | ||||
|
|
||||||||||||||
|
|
Year ended September 30, |
|||||||||||||
|
Condensed Consolidated Balance Sheet Data: |
2016 |
2015 |
2014 |
2013 |
2012 |
|||||||||
|
(In thousands) |
||||||||||||||
|
Cash and cash equivalents |
$ |
2,385 |
$ |
4,106 |
$ |
5,796 |
$ |
8,922 |
$ |
5,296 | ||||
|
Working capital |
14,968 | 17,361 | 9,695 | 13,424 | 10,966 | |||||||||
|
Total assets |
38,624 | 37,472 | 31,673 | 35,170 | 30,446 | |||||||||
|
Accumulated deficit |
(27,651) | (27,996) | (32,342) | (28,715) | (35,594) | |||||||||
|
Long-term obligations |
1,234 | 15 | 39 | 67 | 174 | |||||||||
|
Total stockholders' equity |
$ |
33,933 |
$ |
33,133 |
$ |
28,065 |
$ |
31,403 |
$ |
24,218 | ||||
39
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations
Overview
The Female Health Company is a medical therapeutics company, with an initial focus on the development and commercialization of pharmaceuticals for men’s and women’s health and oncology that qualify for the U.S. Food and Drug Administration's (FDA) 505(b)(2) accelerated regulatory approval pathway. The Company also has a Consumer Health and Medical Devices Division and Global Public Health Sector Division. The Company does business as both "Veru Healthcare" and "The Female Health Company." The Company is organized as follows:
|
· |
Veru Healthcare manages: |
|
o |
The Pharmaceuticals Division, which develops and commercializes pharmaceutical products for men's and women's health and oncology. |
|
o |
The Consumer Health and Medical Devices Division, which is focused on commercializing sexual healthcare products and devices for the consumer market, including the Company’s Female Condom (FC2) for over-the-counter (OTC), and as the Female Disposable Contraceptive Device (FC2) in the U.S. prescription market, as well as PREBOOST® (benzocaine 4%) medicated individual wipes which is a male genital desensitizing drug product that helps in the prevention of premature ejaculation. |
|
· |
The Female Health Company manages the Global Public Health Sector Division, which is focused on the FC2 Female Condom® in the global public health sector business. This division markets FC2 to public health entities, including ministries of health, government health agencies, U.N. agencies, nonprofit organizations and commercial partners, that work to support and improve the lives, health and well-being of women around the world. |
On October 31, 2016, as part of the Company's strategy to diversify its product line to mitigate the risks of being a single product company, the Company completed a merger transaction (the APP Merger) with Aspen Park Pharmaceuticals, Inc. (APP). APP is a medical therapeutics company focused on the development and commercialization of pharmaceutical and consumer health products for men's and women's health and oncology. For men, product and product candidates are in the areas of benign prostatic hyperplasia, male infertility, amelioration of side effects of hormonal prostate cancer therapies, gout, sexual dysfunction, and prostate cancer. For women, product candidates are for advanced breast and ovarian cancers and for female sexual health. APP was originally formed on June 9, 2014, has not had significant revenues and has incurred losses since inception.
On August 12, 2016, the FDA agreed that the Company's Tamsulosin DRS product, a proprietary medication for the treatment of benign prostatic hyperplasia (BPH), a $3.5 billion market, qualifies for the accelerated 505(b)(2) regulatory approval pathway and with APP's plans to conduct a single bioequivalence study to support the filing of a new drug application (NDA). The Company plans to initiate a bioequivalence clinical study by the first quarter of 2017, submit an NDA for Tamsulosin DRS in 2017 and, if approved, launch the product in early 2018.
On October 31, 2016, the Company completed an interim analysis of the double-blind, randomized placebo controlled clinical trial of its novel PREBOOST® product. The Company plans to launch PREBOOST® in the United States before the end of 2016.
The Company accepted an invitation from the FDA to present at the meeting of the Bone, Reproductive and Urologic Drugs (BRUD) Advisory Committee on December 6, 2016. The FDA uses advisory committees to obtain independent expert advice on scientific, technical and policy matters. At the meeting, the committee discussed appropriate clinical trial design features, including acceptable endpoints for demonstrating clinical benefit, for drugs intended to treat secondary hypogonadism (low testosterone levels) while preserving or improving testicular function, including spermatogenesis. At the meeting, the FDA Advisory Committee provided guidance for clinical trial design and endpoints. The committee agreed with the intended patient population to treat, recommended a short-term study, and supported the use of improvement of semen quality for such clinical endpoints as avoidance of aggressive assisted reproductive procedures such as in vitro fertilization or pregnancy. Based on this advice, the Company plans to file an investigational new drug application (IND) in 2017 and advance MSS-722 into Phase 2 clinical trial in men with testicular dysfunction [severe oligospermia (low sperm count) and secondary hypogonadism] as a cause of male factor infertility.
Prior to the completion of the APP Merger, the Company had been a single product company, focused on manufacturing, marketing and selling the Female Condom (FC2). FC2 is the only currently available female-controlled product approved for market by the FDA and cleared by the World Health Organization (WHO) for purchase by U.N. agencies that provides dual protection against unintended pregnancy and sexually transmitted infections (STIs), including HIV/AIDS and the Zika virus.
FC2’s primary use is for disease prevention and family planning, and the public health sector is the Company’s main market. Within the public health sector, various organizations supply critical products such as FC2, at no cost or low cost, to those who need but cannot afford to buy such products for themselves.
40
FC2 has been distributed in 144 countries. A significant number of countries with the highest demand potential are in the developing world. The incidence of HIV/AIDS, other STIs and unwanted pregnancy in these countries represents a remarkable potential for significant sales of a product that benefits some of the world’s most underprivileged people. However, conditions in these countries can be volatile and result in unpredictable delays in program development, tender applications and processing orders.
FC2 has a relatively small customer base, with a limited number of customers who generally purchase in large quantities. Over the past few years, major customers have included large global agencies, such as UNFPA and USAID. Other customers include ministries of health or other governmental agencies, which either purchase directly or via in-country distributors, and NGOs.
Purchasing patterns for FC2 vary significantly from one customer to another, and may reflect factors other than simple demand. For example, some governmental agencies purchase FC2 through a formal procurement process in which a tender (request for bid) is issued for either a specific or a maximum unit quantity. Tenders also define the other elements required for a qualified bid submission (such as product specifications, regulatory approvals, clearance by WHO, unit pricing and delivery timetable). Bidders have a limited period of time in which to submit bids. Bids are subjected to an evaluation process which is intended to conclude with a tender award to the successful bidder. The entire tender process, from publication to award, may take many months to complete. A tender award indicates acceptance of the bidder’s price rather than an order or guarantee of the purchase of any minimum number of units. Many governmental tenders are stated to be “up to” the maximum number of units, which gives the applicable government agency discretion to purchase less than the full maximum tender amount. Orders are placed after the tender is awarded; there are often no set dates for orders in the tender and there are no guarantees as to the timing or amount of actual orders or shipments. Orders received may vary from the amount of the tender award based on a number of factors including vendor supply capacity, quality inspections and changes in demand. Administrative issues, politics, bureaucracy, process errors, changes in leadership, funding priorities and/or other pressures may delay or derail the process and affect the purchasing patterns of public sector customers. As a result, the Company may experience significant quarter-to-quarter sales variations due to the timing and shipment of large orders of FC2.
During fiscal 2011, the Company’s unit shipments, revenues, and net income were adversely affected by bureaucratic delays and other timing issues involving the receipt and shipment of large orders from Brazil and RSA. Significant orders for both countries were received in the first quarter of fiscal 2012. The 20 million unit order received for shipment to Brazil which had been the largest order in the Company’s history. Receipt of these orders positively impacted fiscal 2012 and 2013 results.
In October 2014, the Company announced that Semina was awarded an exclusive contract under a public tender. The contract was valid through August 20, 2015, allowing the Brazil Ministry of Health to place orders against this tender at its discretion. Through the end of the contract, the Company received orders for 40 million units of FC2 in fulfillment of the tender, 28 million of which were shipped during the year ended September 30, 2015 and 12 million of which were shipped during the year ended September 30, 2016.
Details of the quarterly unit sales of FC2 for the last five fiscal years are as follows:
|
Period |
2016 |
2015 |
2014 |
2013 |
2012 |
|
October 1 – December 31 |
15,380,240 | 12,154,570 | 11,832,666 | 17,114,630 | 15,166,217 |
|
January 1 – March 31 |
9,163,855 | 20,760,519 | 7,298,968 | 16,675,035 | 13,945,320 |
|
April 1 – June 30 |
10,749,860 | 14,413,032 | 13,693,652 | 12,583,460 | 15,198,960 |
|
July 1 - September 30 |
6,690,080 | 13,687,462 | 9,697,341 | 8,386,800 | 17,339,500 |
|
Total |
41,984,035 | 61,015,583 | 42,522,627 | 54,759,925 | 61,649,997 |
Revenues. The Company's revenues have been derived from sales of FC2, and are recognized upon shipment of the product to its customers.
The Company is working to further develop a global market and distribution network for FC2 by maintaining relationships with public health sector groups and completing partnership arrangements with companies with the necessary marketing and financial resources and local market expertise.
41
The Company’s most significant customers have been either global public health sector agencies or those who facilitate their purchases and/or distribution of FC2 for use in HIV/AIDS prevention and/or family planning. The Company's four largest customers currently are UNFPA, USAID, Sekunjalo and Semina. UNFPA accounted for 25 percent of unit sales in fiscal 2016, 18 percent of unit sales in fiscal 2015, and 40 percent of unit sales in fiscal 2014. USAID accounted for 24 percent of unit sales in fiscal 2016, 16 percent of unit sales in fiscal 2015, and 17 percent of unit sales in fiscal 2014. Sekunjalo accounted for 13 percent of unit sales in fiscal 2014. Semina accounted for 27 percent of unit sales in fiscal 2016, 47 percent of unit sales in fiscal 2015, and less than 10 percent of unit sales in fiscal 2014. Azinor accounted for 11 percent of unit sales in fiscal 2014. No other single customer accounted for more than 10 percent of unit sales in fiscal 2016, 2015, or 2014. We sell to the Brazil Ministry of Health either through UNFPA or Semina. In the U.S., FC2 is sold to city and state public health clinics as well as to not-for-profit organizations such as Planned Parenthood.
Because the Company manufactures FC2 in a leased facility located in Malaysia, a portion of the Company's operating costs are denominated in foreign currencies. While a material portion of the Company's future sales are likely to be in foreign markets, all sales are denominated in the U.S. dollar. Effective October 1, 2009, the Company’s U.K. and Malaysia subsidiaries adopted the U.S. dollar as their functional currency, further reducing the Company’s foreign currency risk.
Expenses. The Company manufactures FC2 at its facility located in Selangor D.E., Malaysia. The Company's cost of sales consists primarily of direct material costs, direct labor costs and indirect production and distribution costs. Direct material costs include raw materials used to make FC2, principally a nitrile polymer. Indirect production costs include logistics, quality control and maintenance expenses, as well as costs for electricity and other utilities. All of the key components for the manufacture of FC2 are essentially available from either multiple sources or multiple locations within a source.
On April 1, 2015, a tariff exemption in Brazil for condoms was eliminated subjecting all shipments of FC2 clearing customs in Brazil on or after that date to a tariff. The Company agreed to share 50 percent of these tariff costs with Semina and recognized the expense as the units were shipped.
The Company's operating expenses include costs for sales, marketing, education and training relating to FC2. During the London Summit, the Company announced a program to support the London Summit's goal to provide contraceptives to an additional 120 million women by 2020. This program includes a plan for the Company to invest up to $14 million over the period from 2013 through 2018 in reproductive health and HIV/AIDS prevention marketing, education and training in collaboration with global agencies. Such investment in marketing, education and training may increase the Company’s operating expenses in future periods, although the Company has not set a specific timetable for any such increased spending. In connection with the London Summit, the Company implemented a volume purchasing incentive program to award major public sector purchasers with FC2 equal to 5 percent of their total annual units purchased, at no-cost. The Company reserved for the no-cost product as a cost of sales, which impacted the Company’s gross margin. Effective January 1, 2015, the Company reduced the unit price to the major public sector purchasers to reflect the 5 percent no-cost product instead of awarding no-cost product.
Fiscal Year Ended September 30, 2016 Compared to Fiscal Year Ended September 30, 2015
Operating Highlights. The Company had net revenues of $22,127,342 during fiscal 2016, compared to $32,604,865 in fiscal 2015. The Company’s fiscal 2016 unit sales were 19 million units, or 31 percent, lower than fiscal 2015. The decrease in unit sales and net revenues is primarily due to 28 million units shipped during fiscal 2015 under the 2014 Brazilian tender, versus 12 million units shipped during fiscal 2016. The average sales price of FC2 decreased 1.4 percent in fiscal 2016 from fiscal 2015. Effective April 1, 2016, the unit price has been reduced for major public sector purchasers.
The Company used cash in operations of $1,714,358 in fiscal 2016 compared to $1,548,697 in fiscal 2015.
The Company had net income of $344,725, or $0.01 per diluted share, in fiscal 2016 compared to net income of $4,346,036, or $0.15 per diluted share, in fiscal 2015.
Results of Operations. The Company had net revenues of $22,127,342 and net income of $344,725, or $0.01 per diluted share, in fiscal 2016, compared to net revenues of $32,604,865 and net income of $4,346,036, or $0.15 per diluted share, in fiscal 2015. Net revenues decreased $10,477,523, or 32 percent, in fiscal 2016 compared to the prior fiscal year. The reduction in net revenues is due to the lower unit sales, change in sales mix, and public sector price adjustment.
Cost of sales decreased $4,857,048, or 36 percent, to $8,777,858 in fiscal 2016 from $13,634,906 in fiscal 2015. The reduction in cost of sales is due to the lower unit sales, reduction of certain costs, and the favorable impact of currency exchange rates.
Gross profit decreased $5,620,475, or 30 percent, to $13,349,484 in fiscal 2016 from $18,969,959 in fiscal 2015. Gross profit as a percentage of net revenues increased to 60 percent in fiscal 2016 from 58 percent in fiscal 2015. The increase in the gross profit margin is primarily due to the reduction of certain costs and the favorable impact of currency exchange rates on cost of sales.
42
Selling, general and administrative expenses decreased $3,471,563, or 29 percent, to $8,660,174 in fiscal 2016 from $12,131,737 in fiscal 2015. The decrease was a result of a reduction in payments due to our Brazilian distributor for marketing and management fees for the 2014 tender, a reduction in employee compensation expense, a reduction in expenses related to a study regarding a potential FC2 consumer program in the U.S., and a reduction in diversification expenses. The diversification expenses were $548,077 in fiscal 2016 compared to $709,462 in fiscal 2015.
Business acquisition expense of $1,482,539 in fiscal 2016 represents costs related to the merger transaction with APP.
Research and development expenses decreased $120,422 to $99,393 in fiscal 2016 from $219,815 in fiscal 2015.
Total operating expenses decreased $2,020,580 to $10,330,972 in fiscal 2016 from $12,351,552 in fiscal 2015.
The Company's operating income decreased $3,599,895 to $3,018,512 in fiscal 2016 from $6,618,407 in fiscal 2015. The decrease is primarily due to decreased net revenues, partially offset by lower operating expenses and improved gross margins.
The Company recorded non-operating expense of $204,596 in fiscal 2016 compared to non-operating income of $68,633 in fiscal 2015. The impact of the foreign currency transactions was a loss of $147,540 in fiscal 2016 compared to a gain of $58,483 in fiscal 2015.
Income tax expense increased $128,187 to $2,469,191 in fiscal 2016 compared to income tax expense of $2,341,004 in fiscal 2015. The effective tax rate for fiscal 2016 and 2015 was 87.7 percent and 35.0 percent, respectively. The increase in the effective tax rate is due to the mix of tax jurisdictions in which the Company recognized income before income taxes, the non-deductible business acquisition expenses related to the merger transaction with APP, and the reduction in the UK income tax rate from 20% to 18%. The Company’s net operating loss (NOL) carryforwards will be utilized to reduce cash payments for income taxes based on the statutory rate in effect at the time of such utilization. Actual income taxes paid are reflected on the Company’s consolidated statements of cash flows. In fiscal 2016 the Company recorded income tax expense of $2,469,191, while due to the use of NOL carryforwards the Company made cash payments of $352,856 for income taxes.
Fiscal Year Ended September 30, 2015 Compared to Fiscal Year Ended September 30, 2014
Operating Highlights. The Company had net revenues of $32,604,865 during fiscal 2015, compared to $24,490,586 in fiscal 2014. The Company’s fiscal 2015 unit sales were 43 percent higher than fiscal 2014. The increase in unit sales and net revenues is primarily due to 28 million units shipped during fiscal 2015 under the 2014 Brazilian tender. The average sales price of FC2 decreased 7.2 percent in fiscal 2015 from fiscal 2014. Effective January 1, 2015, the unit price has been reduced for major public sector purchasers to replace the previous 5 percent no-cost product policy under the Company’s volume purchasing incentive program. The remaining decrease is primarily due to sales mix.
The Company used cash in operations of $1,548,697 in fiscal 2015 compared to $3,665,413 of cash generated from operations in fiscal 2014.
The Company had net income of $4,346,036, or $0.15 per diluted share, in fiscal 2015 compared to net income of $2,433,061, or $0.08 per diluted share, in fiscal 2014.
Results of Operations. The Company had net revenues of $32,604,865 and net income of $4,346,036, or $0.15 per diluted share, in fiscal 2015, compared to net revenues of $24,490,586 and net income of $2,433,061, or $0.08 per diluted share, in fiscal 2014. Net revenues increased $8,114,279, or 33 percent, in fiscal 2015 compared to the prior fiscal year.
Cost of sales increased $2,265,798, or 20 percent, to $13,634,906 in fiscal 2015 from $11,369,108 in fiscal 2014.
Gross profit increased $5,848,481, or 45 percent, to $18,969,959 in fiscal 2015 from $13,121,478 in fiscal 2014. Gross profit as a percentage of net revenues increased to 58 percent in fiscal 2015 from 54 percent in fiscal 2014. The increase reflects the favorable impact of changes in currency exchange rates slightly offset by higher costs associated with the increased unit sales.
Selling, general and administrative expenses increased $2,997,867, or 33 percent, to $12,131,737 in fiscal 2015 from $9,133,870 in fiscal 2014. Approximately 56 percent of the increased spending related to payments to our Brazilian distributor for ongoing programming related to the 2012 tender and for marketing and management fees related to the 2014 tender. $398,000 of the increase was for the Company’s share of tariff cost related expenses for the Brazilian tender. An accrual for fiscal year end incentive compensation, not incurred in the prior year period, was approximately 15 percent of the increase. Business development consulting costs associated with the portfolio diversification strategy was approximately 23 percent of the increase, minimal costs were incurred in the prior year period. These increased expenses were partially offset by a reduction of expenses relating to employee compensation.
43
Research and development expenses increased $214,240 to $219,815 in fiscal 2015 from $5,575 in fiscal 2014. The increase is primarily related to product enhancements.
Total operating expenses increased $3,153,986 to $12,351,552 in fiscal 2015 from $9,197,566 in fiscal 2014.
The Company's operating income increased $2,694,495, or 69 percent, to $6,618,407 in fiscal 2015 from $3,923,912 in fiscal 2014. The increase is primarily due to increased net revenues and improved gross margins partially offset by higher operating expenses.
The Company recorded non-operating income of $68,633 in fiscal 2015 compared to $33,279 in fiscal 2014. The impact of the foreign currency transactions was a gain of $58,483 in fiscal 2015 compared to a loss of $83,844 in fiscal 2014.
Income tax expense increased $816,874 to $2,341,004 in fiscal 2015 compared to income tax expense of $1,524,130 in fiscal 2014. The effective tax rate for fiscal 2015 and 2014 was 35.0 percent and 38.5 percent, respectively. The reduction in the effective tax rate is due to the mix of tax jurisdictions in which the Company recognized income before income taxes and the reduction in the Illinois state income tax rate, effective January 1, 2015, from 9.5 percent to 7.75 percent. The Company’s net operating loss (NOL) carryforwards will be utilized to reduce cash payments for income taxes based on the statutory rate in effect at the time of such utilization. Actual income taxes paid are reflected on the Company’s consolidated statements of cash flows. In fiscal 2015 the Company recorded income tax expense of $2,341,004, while due to the use of NOL carryforwards the Company made cash payments of $294,441 for income taxes.
Liquidity and Sources of Capital
We have generally funded our operations and working capital needs through cash generated from operations. Our operating activities used cash of $1.7 million in fiscal 2016, used cash of $1.5 million in fiscal 2015, and generated cash of $3.7 million in fiscal 2014. Accounts receivable and long-term other receivables increased from $14.1 million at September 30, 2015 to $18.6 million at September 30, 2016. Semina’s accounts receivable and long-term other receivables balance represents 85 percent of the Company’s accounts receivable and long-term other receivables balance at September 30, 2016. Semina normally pays upon payment from the Brazilian Government; however due to economic issues in Brazil the government has been slower in paying vendors. In addition, total current liabilities decreased $0.9 million, primarily due to $1.2 million owed to Semina related to the 2014 tender moving from current liabilities to long-term liabilities. In fiscal 2016, investing activities used cash of $6,374 and there were no financing activities. In fiscal 2015, investing activities used cash of $135,424 and financing activities used cash of $6,288.
At September 30, 2016, the Company had working capital of $15.0 million and stockholders' equity of $33.9 million compared to working capital of $17.4 million and stockholders' equity of $33.1 million as of September 30, 2015.
Beginning February 16, 2010, through May 7, 2014, the Company paid a total of 18 consecutive dividends. The first 9 quarterly dividends were paid at a quarterly rate per share of $0.05 through February 9, 2012, 4 were paid at a quarterly rate per share of $0.06 from May 9, 2012 through February 6, 2013 and 5 were paid at a quarterly rate per share of $0.07 from May 8, 2013 through May 7, 2014. Cash dividends paid totaled $29.4 million during this period. The Company paid cash dividends of approximately $6.1 million and $7.5 million in fiscal 2014 and fiscal 2013, respectively. On July 14, 2014, the Company announced that its Board of Directors has elected to suspend the payment of quarterly cash dividends in order to devote operating cash flows towards strategic growth initiatives.
The Company believes its current cash position is adequate to fund operations of the Company in the next 12 months, although no assurances can be made that such cash will be adequate. Depending on the timing of payment of the Company's outstanding accounts receivable and long-term other receivables balance due from Semina and the timing of development activities relating to the Company's drug candidates, the Company may decide to raise additional capital in the near term. If the Company needs additional cash, potential sources of such cash would include the sale of equity, convertible debt or other equity-linked securities or the borrowing of funds under its BMO Harris Bank N.A. credit facility.
44
On December 29, 2015, the Company entered into a Credit Agreement (the Credit Agreement) with BMO Harris Bank N.A. (BMO Harris Bank). The Credit Agreement provides the Company with a revolving line of credit of up to $10 million with a term that extends to December 29, 2017. Borrowings under the Credit Agreement bear interest, at the Company’s option, at a base rate or at LIBOR plus 2.25%. The Company is also required to pay a commitment fee at the rate of 0.10% per annum on the average daily unused portion of the revolving line of credit. The Company's obligations under the Credit Agreement are secured by a lien against substantially all of the assets of the Company and a pledge of 65% of the outstanding shares of The Female Health Company Limited. In addition to other customary representations, covenants and default provisions, the Company is required to maintain a minimum tangible net worth and to not to exceed a maximum total leverage ratio. Among the non-financial covenants, the Company is restricted in its ability to pay dividends, buy back shares of its common stock, incur additional debt and make acquisitions. No amounts were outstanding under the Credit Agreement at September 30, 2016.
The completion of the APP Merger resulted in a default in FHC's compliance with certain covenants in the Credit Agreement and will constitute an "event of default" under the Credit Agreement. On November 28, 2016, FHC, Badger Acquisition Sub, Inc., APP and BMO Harris Bank entered into a Third Amendment to the Credit Agreement (the "Amendment"). Pursuant to the Amendment, BMO Harris Bank waived the defaults in FHC's compliance with the covenants in the Credit Agreement as a result of the completion of the APP Merger and APP became a co-borrower under the Credit Agreement. As a result, the revolving line of credit remains in effect under the terms of the Credit Agreement until the end of its term on December 29, 2017.
As of December 9, 2016, the Company had approximately $1.6 million in cash, net trade accounts receivable of $17.6 million and current trade accounts payable of $0.8 million. Presently, the Company has no required debt service obligations.
The following table includes information relating to our contractual obligations as of September 30, 2016 in future fiscal years:
|
|
||||||||||||||||||||
|
|
||||||||||||||||||||
|
Contractual |
||||||||||||||||||||
|
Obligations |
Total |
2017 |
2018 |
2019 |
2020 |
2010 |
Thereafter |
|||||||||||||
|
Long-term debt |
$ |
- |
$ |
- |
$ |
- |
$ |
- |
$ |
- |
$ |
- |
$ |
- |
||||||
|
Capital lease obligations |
- |
- |
- |
- |
- |
- |
- |
|||||||||||||
|
Operating lease obligations |
1,674,235 | 345,301 | 401,624 | 401,904 | 186,305 | 110,803 | 228,298 | |||||||||||||
|
Purchase obligations |
- |
- |
- |
- |
- |
- |
- |
|||||||||||||
|
Other long-term obligations |
1,233,750 |
- |
1,233,750 |
- |
- |
- |
- |
|||||||||||||
|
Total |
$ |
2,907,985 |
$ |
345,301 |
$ |
1,635,374 |
$ |
401,904 |
$ |
186,305 |
$ |
110,803 |
$ |
228,298 | ||||||
Critical Accounting Estimates
The preparation of financial statements requires management to make estimates and use assumptions that affect certain reported amounts and disclosures. Critical accounting estimates include the deferred income tax valuation allowance. Actual results may differ from those estimates.
The Company files separate income tax returns for its foreign subsidiaries. ASC Topic 740 requires recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial statements and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Deferred tax assets are also provided for carryforwards for income tax purposes. In addition, the amount of any future tax benefits is reduced by a valuation allowance to the extent such benefits are not expected to be realized.
The Company accounts for income taxes using the liability method, which requires the recognition of deferred tax assets or liabilities for the tax-effected temporary differences between the financial reporting and tax bases of assets and liabilities, and for net operating loss and tax credit carryforwards.
45
The Company completes a detailed analysis of its deferred income tax valuation allowance on an annual basis or more frequently if information comes to our attention that would indicate that a revision to its estimates is necessary. In evaluating the Company’s ability to realize its deferred tax assets, management considers all available positive and negative evidence on a country by country basis, including past operating results and forecast of future taxable income. In determining future taxable income, management makes assumptions to forecast U.S. federal and state, U.K. and Malaysia operating income, the reversal of temporary differences, and the implementation of any feasible and prudent tax planning strategies. These assumptions require significant judgment regarding the forecasts of the future taxable income in each tax jurisdiction, and are consistent with the forecasts used to manage the Company’s business. It should be noted that the Company realized significant losses through 2005 on a consolidated basis. Since fiscal 2006, the Company has consistently generated taxable income on a consolidated basis, providing a reasonable future period in which the Company can reasonably expect to generate taxable income. In management’s analysis to determine the amount of the deferred tax asset to recognize, management projected future taxable income for each tax jurisdiction.
Although management uses the best information available, it is reasonably possible that the estimates used by the Company will be materially different from the actual results. These differences could have a material effect on the Company's future results of operations and financial condition.
Our effective tax rates have differed from the statutory rate primarily due to the tax impact of foreign operations, state taxes and reversal of the valuation allowance against the NOL carryforwards. Our future effective tax rates could be adversely affected by earnings being lower than anticipated in countries where we have lower statutory rates and higher than anticipated in countries where we have higher statutory rates, changes in the valuation of our deferred tax assets or liabilities, or changes in tax laws, regulations, and accounting principles. In addition, we are subject to the continuous examination of our income tax returns by the IRS and other tax authorities. We regularly assess the likelihood of adverse outcomes resulting from these examinations to determine the adequacy of our provision for income taxes.
Impact of Inflation and Changing Prices
Although the Company cannot accurately determine the precise effect of inflation, the Company has experienced increased costs of product, supplies, salaries and benefits, and increased general and administrative expenses. The Company has, where possible, increased selling prices to offset such increases in costs.
Off-Balance Sheet Arrangements
The Company has no off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
46
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
The Company's exposure to market risk is limited to fluctuations in raw material commodity prices, particularly the nitrile polymer used to manufacture FC2, and foreign currency exchange rate risk associated with the Company's foreign operations. The Company does not utilize financial instruments for trading purposes or to hedge risk and holds no derivative financial instruments which would expose it to significant market risk. Effective October 1, 2009, the Company's U.K. subsidiary and Malaysia subsidiary each adopted the U.S. dollar as its functional currency. The consistent use of the U.S. dollar as the functional currency across the Company reduces its foreign currency risk and stabilizes its operating results. The Company’s distributors are subject to exchange rate risk as their orders are denominated in the U.S. dollars and they generally sell to their customers in the local country currency. If currency fluctuations have a material impact on a distributor it may ask the Company for pricing concessions or other financial accommodations. The Company currently has no significant exposure to interest rate risk. The Company has a line of credit with BMO Harris Bank, consisting of a revolving note for up to $10 million. Outstanding borrowings under the line of credit will incur interest, at the Company’s option, at a base rate or at LIBOR plus 2.25%. As the Company has had no outstanding borrowings in the last five years, it currently has no significant exposure to market risk for changes in interest rates. Should the Company incur future borrowings under its line of credit, it would be subject to interest rate risk related to such borrowings.
Item 8. Financial Statements and Supplementary Data
The response to this item is submitted in a separate section of this report. See “Index to Consolidated Financial Statements” for a list of the financial statements being filed herein.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
As of the end of the period covered by this report, the Company carried out an evaluation, under the supervision and with the participation of the Company's management, including the Company's Chief Executive Officer and the Company's Chief Financial Officer, of the effectiveness of the design and operation of the Company's disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended). Based on this evaluation, the Company's Chief Executive Officer and Chief Financial Officer concluded that the Company's disclosure controls and procedures were effective. It should be noted that in designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. The Company has designed its disclosure controls and procedures to reach a level of reasonable assurance of achieving desired control objectives and, based on the evaluation described above, the Company's Chief Executive Officer and Chief Financial Officer concluded that the Company's disclosure controls and procedures were effective at reaching that level of reasonable assurance.
Changes in Internal Control Over Financial Reporting
There was no change in the Company's internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended) during the Company's most recently completed fiscal quarter that has materially affected, or is reasonably likely to materially affect, the Company's internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
The report of management required under this Item 9A is contained on page F-1 of this Annual Report on Form 10-K under the heading "Management's Report on Internal Control over Financial Reporting."
Report of Independent Registered Public Accounting Firm
The attestation report required under this Item 9A is contained on page F-2 of this Annual Report on Form 10-K under the heading "Report of Independent Registered Public Accounting Firm."
None.
47
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Information with respect to this item is incorporated herein by reference to the discussion under the headings “Proposal 1: Election of Directors,” “Executive Officers,” “Section 16(a) Beneficial Ownership Reporting Compliance,” “Corporate Governance Matters-Director Nominations” and “Audit Committee Matters – Audit Committee Financial Expert” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017. Information regarding the Company’s Code of Business Ethics is incorporated herein by reference to the discussion under “Corporate Governance Matters –Code of Business Ethics” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017.
The Audit Committee of the Company’s Board of Directors is an “audit committee” for purposes of Section 3(a)(58)(A) of the Securities Exchange Act of 1934. The members of the Audit Committee are Mary Margaret Frank (Chairperson), Lucy Lu and Georges Makhoul.
Item 11. Executive Compensation
Information with respect to this item is incorporated herein by reference to the discussion under the headings “Director Compensation and Benefits,” and “Executive Compensation” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017. The information under the subsection "Executive Compensation – Compensation Committee Report" is not deemed to be "soliciting material" or to be "filed" with the SEC or subject to Regulation 14A under the Securities Exchange Act of 1934 or to be the liabilities of Section 18 of the Securities Exchange Act of 1934, and will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except to the extent it is specifically incorporated by reference into such a filing.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information with respect to this item is incorporated herein by reference to the discussion under the heading “Security Ownership” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017.
Equity Compensation Plan Information
The following table summarizes share information, as of September 30, 2016, for the Company's equity compensation plans and arrangements. The plans and arrangements dated prior to July 2007 were not required to be approved by the Company's shareholders, and, accordingly, none of these plans or arrangements have been approved by the Company's shareholders. In March 2008, the Company’s shareholders approved the 2008 Stock Incentive Plan and authorized 2 million shares (subject to adjustment in the event of stock splits and other similar events) for issuance under the plan.
|
|
||||||
|
|
||||||
|
|
Shares Remaining |
|||||
|
|
Number of Shares To Be |
Weighted-Average |
Available For Future |
|||
|
|
Issued Upon Exercise Of |
Exercise Price Of |
Issuance Under Equity |
|||
|
Equity Plan Category |
Outstanding Options |
Outstanding Options |
Compensation Plans |
|||
|
Equity compensation plans approved by shareholders |
191,999 |
(1) |
$ |
2.54 | 528,698 | |
|
Equity compensation plans not approved by shareholders |
90,000 |
$ |
1.27 |
— |
||
|
Total |
281,999 |
$ |
2.14 | 528,698 | ||
______________
(1)Includes a right to receive a total of 84,499 shares, or at a holder’s election cash based on the fair market value of the shares, contingent on continued employment or service.
48
The Company's equity compensation plans not approved by shareholders consists of the 1997 Stock Option Plan. Options granted under the 1997 Stock Option Plan are nonqualified stock options under the Internal Revenue Code. Options expire at such time as the Board of Directors determines, provided that no stock option may be exercised later than the tenth anniversary of the date of its grant. Options cannot be exercised until the vesting period, if any, specified by the Board of Directors. Options are not transferable other than by will or the laws of descent and distribution, and may be exercised during the life of the participant only by him or her. The option price per share is determined by the Board of Directors, but cannot be less than 100 percent of the fair market value of the common stock on the date such option is granted. The 1997 Stock Option Plan expired as of December 31, 2006, thus no further shares can be issued under this plan.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Information with respect to this item is incorporated herein by reference to the discussion under the heading “Certain Relationships and Related Transactions” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017. Information regarding director independence is incorporated by reference to the discussion under “Corporate Governance Matters – Director Independence” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017.
Item 14. Principal Accountant Fees and Services.
Information with respect to this item is incorporated herein by reference to the discussion under the heading “Audit Committee Matters – Fees of Independent Registered Public Accounting Firm” in the Company’s Proxy Statement for the 2017 Annual Meeting of Shareholders, which will be filed with the SEC on or before January 27, 2017.
49
PART IV
Item 15. Exhibits and Financial Statement Schedules.
(a) The following documents are filed as part of this report:
1.Financial Statements
|
|
The following consolidated financial statements of the Company are included in Item 8 of this report: |
|
|
Report of Independent Registered Public Accounting Firm |
|
|
Consolidated Balance Sheets as of September 30, 2016 and 2015 |
|
|
|
|
|
Consolidated Statements of Income for the Years Ended September 30, 2016, 2015, and 2014 |
|
|
Consolidated Statements of Stockholders’ Equity for the Years Ended September 30, 2016, 2015, and 2014 |
|
|
Consolidated Statements of Cash Flows for the Years Ended September 30, 2016, 2015, and 2014 |
|
|
Notes to Consolidated Financial Statements |
2.Financial Statement Schedules
All schedules for which provision is made in the applicable accounting regulations of the SEC are not required under the related instructions, are inapplicable or the required information is shown in the financial statements or notes thereto, and therefore, have been omitted.
50
3.Exhibits
|
2.1 |
Amended and Restated Agreement and Plan of Merger, dated as of October 31, 2016, among the Company, Blue Hen Acquisition, Inc. and APP. (1) |
|
|
|
|
3.1 |
Amended and Restated Articles of Incorporation of the Company. (2) |
|
|
|
|
3.2 |
Articles of Amendment to the Amended and Restated Articles of Incorporation of the Company increasing the number of authorized shares of common stock to 27,000,000 shares. (3) |
|
|
|
|
3.3 |
Articles of Amendment to the Amended and Restated Articles of Incorporation of the Company increasing the number of authorized shares of common stock to 35,500,000 shares. (4) |
|
|
|
|
3.4 |
Articles of Amendment to the Amended and Restated Articles of Incorporation of the Company increasing the number of authorized shares of common stock to 38,500,000 shares. (5) |
|
|
|
|
3.5 |
Articles of Amendment to the Amended and Restated Articles of Incorporation of the Company designating the terms and preferences for the Class A Preferred Stock – Series 3. (6) |
|
|
|
|
3.6 |
Articles of Amendment to the Amended and Restated Articles of Incorporation of the Company designating the terms and preferences for the Class A Preferred Stock – Series 4. (1) |
|
|
|
|
3.7 |
Amended and Restated By-Laws of the Company. (7) |
|
|
|
|
4.1 |
Amended and Restated Articles of Incorporation, as amended (same as Exhibits 3.1, 3.2, 3.3, 3.4, 3.5 and 3.6). |
|
|
|
|
4.2 |
Articles II, VII and XI of the Amended and Restated By-Laws of the Company (included in Exhibit 3.7). |
|
|
|
|
10.1 |
Form of Lock-Up Agreement, dated as of October 31, 2016, between the Company and each of Mitchell S. Steiner M.D., Harry Fisch, M.D. and K&H Fisch Family Partners LLC. (1) |
|
|
|
|
10.2 |
Registration Rights Agreement, dated as of October 31, 2016, among the Company and the former stockholders of APP. (1) |
|
|
|
|
10.3 |
Escrow Agreement, dated as of October 31, 2016, among the Company, O.B. Parrish, David R. Bethune and Mary Margaret Frank, Ph.D., acting as the committee representing the interests of the Company, Mitchell S. Steiner, M.D., in his capacity as nominee for the stockholders of the Company identified on Exhibit A thereto, and Computershare Trust Company, N.A., as escrow agent. (1) |
|
|
|
|
10.4 |
Warrant to Purchase Common Stock, dated October 31, 2016, issued by the Company to Torreya Capital, a division of Financial West Investment Group. (1) |
|
|
|
|
10.5 |
Separation Agreement and General Release, dated as of July 10, 2015, among the Company, Karen King and certain directors of the Company. (8) |
|
|
|
|
10.6 |
Employment Agreement, dated April 5, 2016, between the Company and Mitchell S. Steiner, M.D. (9) |
|
|
|
|
10.7 |
First Amendment to Employment Agreement, dated as of July 18, 2016, between the Company and Mitchell S. Steiner, M.D. |
|
|
|
|
10.8 |
Employment Agreement, dated April 5, 2016, between the Company and Michele Greco. (9) |
|
|
|
|
10.9 |
First Amendment to Employment Agreement, dated as of July 18, 2016, between the Company and Michele Greco. |
|
|
|
|
10.10 |
Employment Agreement, dated April 5, 2016, between the Company and Martin Tayler. (9) |
|
|
|
|
10.11 |
First Amendment to Employment Agreement, dated as of July 18, 2016, between the Company and Martin Tayler. |
|
|
|
|
10.12 |
The Female Health Company 2008 Stock Incentive Plan. (10) |
|
|
|
|
10.13 |
Form of Nonstatutory Stock Option Grant Agreement for The Female Health Company 2008 Stock Incentive Plan. (11) |
|
|
|
|
10.14 |
Form of Restricted Stock Grant Agreement for The Female Health Company 2008 Stock Incentive Plan. (12) |
51
|
|
|
|
10.15 |
Credit Agreement, dated as of December 29, 2015, between the Company and BMO Harris Bank N.A. (13) |
|
|
|
|
10.16 |
First Amendment and Waiver to Credit Agreement and Security Agreement, dated as of January 4, 2016, between the Company and BMO Harris Bank N.A. |
|
|
|
|
10.17 |
Consent and Amendment to Credit Agreement, dated as of March 31, 2016, between the Company and BMO Harris Bank N.A. (14) |
|
|
|
|
10.18 |
Revolving Note, dated December 29, 2015, from the Company to BMO Harris Bank N.A. (13) |
|
|
|
|
10.19 |
Security Agreement, dated as of December 29, 2015, between the Company and BMO Harris Bank N.A. (13) |
|
|
|
|
10.20 |
Charge Over Shares Agreement, dated as of December 29, 2015, between The Female Health Company and BMO Harris Bank N.A. (13) |
|
|
|
|
10.21 |
Second Amendment to Security Agreement and First Amendment to Subsidiary Security Agreement, dated as of September 29, 2016, between the Company and BMO Harris Bank N.A. |
|
|
|
|
21 |
Subsidiaries of Registrant. |
|
|
|
|
23.1 |
Consent of RSM US LLP. |
|
|
|
|
24.1 |
Power of Attorney (included as part of the signature page hereof). |
|
|
|
|
31.1 |
Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
31.2 |
Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
32.1 |
Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 (Section 906 of the Sarbanes-Oxley Act of 2002. (15) |
|
|
|
|
101 |
The following materials from the Company's Annual Report on Form 10-K for the year ended September 30, 2016, formatted in XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Income, (iii) Consolidated Statements of Stockholders’ Equity, (iv) Consolidated Statements of Cash Flows, and (v) the Notes to Consolidated Financial Statements. |
___________
|
(1) |
Incorporated herein by reference to the Company's Form 8-K filed on November 2, 2016. |
|
|
|
|
(2) |
Incorporated herein by reference to the Company's Form SB-2 Registration Statement filed on October 19, 1999. |
|
|
|
|
(3) |
Incorporated herein by reference to the Company's Form SB-2 Registration Statement filed on September 21, 2000. |
|
|
|
|
(4) |
Incorporated herein by reference to the Company's Form SB-2 Registration Statement filed on September 6, 2002. |
|
|
|
|
(5) |
Incorporated herein by reference to the Company's March 31, 2003 Form 10-QSB. |
|
|
|
|
(6) |
Incorporated herein by reference to the Company's March 31, 2004 Form 10-QSB. |
|
|
|
|
(7) |
Incorporated herein by reference to the Company's Form 8-K filed on May 22, 2013. |
|
|
|
|
(8) |
Incorporated by reference to the Company’s Form 8-K filed on July 16, 2015. |
|
|
|
|
(9) |
Incorporated herein by reference to the Company's Form 8-K filed on April 6, 2016. |
|
|
|
|
(10) |
Incorporated herein by reference to the Company's Form 8-K filed on March 31, 2008. |
|
|
|
|
(11) |
Incorporated herein by reference to the Company's September 30, 2009 Form 10-K. |
|
|
|
52
|
(12) |
Incorporated herein by reference to the Company's September 30, 2013 Form 10-K. |
|
|
|
|
(13) |
Incorporated herein by reference to the Company's Form 8-K filed on January 4, 2016. |
|
|
|
|
(14) |
Incorporated herein by reference to the Company's June 30, 2016 Form 10-Q. |
|
|
|
|
(15) |
This certification is not "filed" for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or incorporated by reference into any filing under the Securities Exchange Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended. |
|
|
|
The response to this portion of Item 15 is submitted as a separate section of this report.
(c) Financial Statement Schedules
53
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Date: December 12, 2016 |
|
THE FEMALE HEALTH COMPANY |
||
|
|
|
|
|
|
|
|
|
BY: |
/s/ Mitchell Steiner |
|
|
|
|
|
Mitchell Steiner, President and |
|
|
|
|
|
Chief Executive Officer |
|
|
|
|
|
|
|
|
|
|
BY: |
/s/ Daniel Haines |
|
|
|
|
|
Daniel Haines, Chief Financial Officer |
|
POWER OF ATTORNEY
Each person whose signature appears below hereby appoints Mitchell Steiner and Daniel Haines, and each of them individually, his true and lawful attorney-in-fact, with power to act with or without the other and with full power of substitution and resubstitution, in any and all capacities, to sign any or all amendments to the Form 10-K and file the same with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or their substitutes, may lawfully cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the date indicated.
|
|
||
|
Signature |
Title |
Date |
|
|
||
|
/s/ Mitchell Steiner |
President, Chief Executive Officer and Director |
December 12, 2016 |
|
Mitchell Steiner |
(Principal Executive Officer) |
|
|
|
||
|
/s/ Daniel Haines |
Chief Financial Officer |
December 12, 2016 |
|
Daniel Haines |
(Principal Accounting and Financial Officer) |
|
|
|
||
|
/s/ Elgar Peerschke |
Chairman of the Board |
December 12, 2016 |
|
Elgar Peerschke |
||
|
|
||
|
/s/ O.B. Parrish |
Vice Chairman of the Board |
December 12, 2016 |
|
O.B. Parrish |
||
|
|
||
|
/s/ David R. Bethune |
Director |
December 12, 2016 |
|
David R. Bethune |
||
|
|
||
|
/s/ Mario Eisenberger |
Director |
December 12, 2016 |
|
Mario Eisenberger |
||
|
|
||
|
/s/ Harry Fisch |
Director |
December 12, 2016 |
|
Harry Fisch |
||
|
|
||
|
/s/ Mary Margaret Frank |
Director |
December 12, 2016 |
|
Mary Margaret Frank |
||
|
|
||
|
/s/ Lucy Lu |
Director |
December 12, 2016 |
|
Lucy Lu |
||
|
|
||
|
/s/ Georges Makhoul |
Director |
December 12, 2016 |
|
Georges Makhoul |
54
Female Health Company
Index to Consolidated Financial Statements
|
Document |
Page No. |
|
Audited Consolidated Financial Statements. |
|
|
Management's Report on Internal Control over Financial Reporting. |
F-1 |
|
Report of RSM US LLP, Independent Registered Public Accounting Firm. |
F-2 |
|
Consolidated Balance Sheets as of September 30, 2016 and 2015. |
F-3 |
|
Consolidated Statements of Income for the years ended September 30, 2016, 2015, and 2014. |
F-4 |
|
F-5 through F-7 |
|
|
Consolidated Statements of Cash Flows for the years ended September 30, 2016, 2015 and 2014. |
F-8 |
|
F-9 through F-24 |